https://scib-metrics.readthedocs.io/en/latest/notebooks/lung_example.html#
Setup#
Check the environment, import dependencies, and set paths.
import sys
print(sys.version)
print(sys.executable)
import importlib
print(importlib.util.find_spec("scbiot"))
import scbiot as scb
3.12.8 (main, Jan 14 2025, 22:49:14) [Clang 19.1.6 ]
/home/figo/software/python_libs/scbiot/.venv/bin/python
ModuleSpec(name='scbiot', loader=<_frozen_importlib_external.SourceFileLoader object at 0x7766ddfb9730>, origin='/home/figo/software/python_libs/scbiot/src/scbiot/__init__.py', submodule_search_locations=['/home/figo/software/python_libs/scbiot/src/scbiot'])
scbiot version 1.1.7
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/_utils/__init__.py:33: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
from anndata import __version__ as anndata_version
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/__init__.py:24: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
if Version(anndata.__version__) >= Version("0.11.0rc2"):
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/readwrite.py:16: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
if Version(anndata.__version__) >= Version("0.11.0rc2"):
import warnings
warnings.filterwarnings("ignore")
import numpy as np
import scanpy as sc
import seaborn as sns
import matplotlib.pyplot as plt
# %pip install torch torchvision torchaudio --index-url https://download.pytorch.org/whl/cu124
import torch
import os
import pandas as pd
import scbiot as scb
from scbiot.utils import set_seed
import harmonypy as hm
from umap import UMAP
# %pip install scib-metrics
from scib_metrics.benchmark import Benchmarker, BioConservation, BatchCorrection
set_seed(42)
from pathlib import Path
dir = Path(os.environ.get("SCBIOT_EXAMPLES_PATH", Path.cwd()))
print(dir)
parent_dir = dir.parent
print(parent_dir)
Random seed set as 42
/home/figo/software/python_libs/scbiot/examples
/home/figo/software/python_libs/scbiot
Load#
Read the lung atlas dataset from disk.
adata_path = f"{dir}/inputs/panc8.h5ad"
adata = sc.read(
adata_path,
# backup_url="https://figshare.com/ndownloader/files/24539942",
)
adata.obs['tech'].value_counts()
tech
indrop 8569
smartseq2 2394
celseq2 2285
celseq 1004
fluidigmc1 638
Name: count, dtype: int64
adata.obs['cell_type'] = adata.obs['celltype']
adata.obs['batch'] = adata.obs['tech']
Preprocess#
Create semi-supervised labels and compute PCA features. Here, we randomly select 20% of cells as truly labeled and assign the remaining cells the label Unknown. For custom datasets, you can instead label high-confidence cells by thresholding marker-gene module scores at the 80th percentile, then propagate these seed labels to the remaining cells with the supBIOT method.
import pandas as pd
def set_reference_batch_labels(
adata,
*,
batch_key="batch",
ref_batch="4", # compare as string to handle mixed int/str batches
label_key="cell_type",
out_key="semi_cell_type",
unlabeled_tag="Unknown",
):
# normalize batch to string for robust matching (your obs['batch'] mixes ints/strings)
b = adata.obs[batch_key].astype(str)
is_ref = (b == str(ref_batch))
# true labels as string; fill NA as Unknown
true_lab = adata.obs[label_key].astype("string").fillna(unlabeled_tag)
# categories: preserve existing category order if categorical, else sorted uniques
if pd.api.types.is_categorical_dtype(adata.obs[label_key]):
cats = list(adata.obs[label_key].cat.categories.astype(str))
else:
cats = sorted(pd.unique(true_lab.astype(str)))
if unlabeled_tag not in cats:
cats.append(unlabeled_tag)
# assign: keep labels in ref batch, mask others
semi = pd.Series(unlabeled_tag, index=adata.obs_names, dtype="string")
semi.loc[is_ref] = true_lab.loc[is_ref].astype("string")
adata.obs[out_key] = pd.Categorical(semi.astype(str), categories=cats)
# quick sanity prints
print(f"[semi labels] ref_batch={ref_batch} labeled={is_ref.sum()} / total={adata.n_obs}")
print(adata.obs[out_key].value_counts().head(20))
print("\n[per-batch labeled fraction]")
tmp = pd.DataFrame({
"batch": b,
"is_labeled": adata.obs[out_key].astype(str).ne(unlabeled_tag),
})
print(tmp.groupby("batch")["is_labeled"].mean().sort_values(ascending=False).head(20))
# --- usage ---
set_reference_batch_labels(adata, batch_key="batch", ref_batch="indrop", label_key="cell_type", out_key="semi_cell_type")
[semi labels] ref_batch=indrop labeled=8569 / total=14890
semi_cell_type
Unknown 6321
beta 2507
alpha 2309
acinar 1152
ductal 915
delta 608
activated_stellate 294
gamma 266
endothelial 235
quiescent_stellate 160
macrophage 55
mast 39
epsilon 16
schwann 13
Name: count, dtype: int64
[per-batch labeled fraction]
batch
indrop 1.0
celseq 0.0
celseq2 0.0
fluidigmc1 0.0
smartseq2 0.0
Name: is_labeled, dtype: float64
# sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
# sc.pp.log1p(adata)
# sc.pp.highly_variable_genes(adata, n_top_genes=2000, flavor="cell_ranger", batch_key='batch')
# sc.pp.scale(adata)
# sc.tl.pca(adata, n_comps=50, use_highly_variable=True)
sc.pp.highly_variable_genes(adata, n_top_genes=2000, flavor="seurat_v3", batch_key='batch')
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.scale(adata)
sc.tl.pca(adata, n_comps=30, use_highly_variable=True)
adata_pre = adata.copy()
Integrate#
Run supervised OT integration and infer labels with supBIOT.
adata, metrics = scb.ot.integrate(
adata,
obsm_key="X_pca",
batch_key="batch",
# prealign='coral',
out_key="X_supbiot",
label_key="semi_cell_type",
unlabeled_category="Unknown",
run_three_stage=True
)
print(metrics)
adata = scb.ot.supbiot(
adata,
label_key="semi_cell_type",
unlabeled_category="Unknown",
pred_label_key="pred_cell_type",
pred_conf_key="pred_confidence",
min_conf=0.0,
)
======== Stage1: supervised OT for label propagation ========
[baseline] KNN backend=FAISS-GPU mix=0.0444 strain=0.00000
[iter 01] mix=0.051 overlap0=0.943 strain=0.00065 floor~0.600 J=0.169 best_it=1
[iter 02] mix=0.060 overlap0=0.902 strain=0.00209 floor~0.607 J=0.175 best_it=2
[iter 03] mix=0.073 overlap0=0.864 strain=0.00458 floor~0.614 J=0.181 best_it=3
[iter 04] mix=0.092 overlap0=0.819 strain=0.00851 floor~0.621 J=0.183 best_it=4
[iter 05] mix=0.121 overlap0=0.773 strain=0.01367 floor~0.629 J=0.196 best_it=5
[iter 06] mix=0.165 overlap0=0.722 strain=0.01970 floor~0.636 J=0.222 best_it=6
[iter 07] mix=0.231 overlap0=0.660 strain=0.02702 floor~0.643 J=0.263 best_it=7
[iter 08] mix=0.318 overlap0=0.597 strain=0.03488 floor~0.650 J=0.325 best_it=8
[iter 09] mix=0.412 overlap0=0.534 strain=0.04153 floor~0.657 J=0.381 best_it=9
[iter 10] mix=0.464 overlap0=0.491 strain=0.04808 floor~0.664 J=0.405 best_it=10
[iter 11] mix=0.483 overlap0=0.458 strain=0.05413 floor~0.671 J=0.393 best_it=10
[iter 12] mix=0.479 overlap0=0.457 strain=0.05523 floor~0.679 J=0.383 best_it=10
[iter 13] mix=0.469 overlap0=0.463 strain=0.05457 floor~0.686 J=0.376 best_it=10
[early stop] plateau reached.
[final] it*=10 mix=0.464 overlap0=0.491 strain=0.04808 tw=0.947
======== Stage2: unsupervised OT for batch integration ========
[baseline] KNN backend=FAISS-GPU mix=0.4635 strain=0.00000
[iter 01] mix=0.479 overlap0=0.924 strain=0.00072 floor~0.600 J=0.166 best_it=1
[iter 02] mix=0.488 overlap0=0.878 strain=0.00280 floor~0.607 J=0.177 best_it=2
[iter 03] mix=0.491 overlap0=0.825 strain=0.00736 floor~0.614 J=0.161 best_it=2
[iter 04] mix=0.494 overlap0=0.833 strain=0.00660 floor~0.621 J=0.171 best_it=2
[iter 05] mix=0.499 overlap0=0.836 strain=0.00531 floor~0.629 J=0.179 best_it=5
[iter 06] mix=0.512 overlap0=0.780 strain=0.01114 floor~0.636 J=0.168 best_it=5
[iter 07] mix=0.494 overlap0=0.778 strain=0.01105 floor~0.643 J=0.150 best_it=5
[iter 08] mix=0.511 overlap0=0.783 strain=0.01053 floor~0.650 J=0.169 best_it=5
[early stop] plateau reached.
[final] it*=5 mix=0.499 overlap0=0.836 strain=0.00531 tw=0.999
[label transfer] skipped; pass label_key to compute alignment metadata
======== Stage3: supervised OT for refinement ========
[baseline] KNN backend=FAISS-GPU mix=0.4995 strain=0.00000
[iter 01] mix=0.499 overlap0=0.927 strain=0.00118 floor~0.600 J=0.151 best_it=1
[iter 02] mix=0.507 overlap0=0.876 strain=0.00452 floor~0.607 J=0.155 best_it=2
[iter 03] mix=0.512 overlap0=0.845 strain=0.00994 floor~0.614 J=0.157 best_it=3
[iter 04] mix=0.520 overlap0=0.809 strain=0.01608 floor~0.621 J=0.150 best_it=3
[iter 05] mix=0.519 overlap0=0.807 strain=0.01665 floor~0.629 J=0.148 best_it=3
[iter 06] mix=0.522 overlap0=0.794 strain=0.01714 floor~0.636 J=0.141 best_it=3
[early stop] plateau reached.
[final] it*=3 mix=0.512 overlap0=0.845 strain=0.00994 tw=0.999
{'mix': 0.5119088351720286, 'overlap0': 0.8445800542831421, 'strain': 0.00994451334526108, 'tw': 0.9993593861614721, 'it': 3}
adata
AnnData object with n_obs × n_vars = 14890 × 34363
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'tech', 'replicate', 'assigned_cluster', 'celltype', 'dataset', 'ident', 'cell_type', 'batch', 'semi_cell_type', 'pred_cell_type', 'pred_confidence'
var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches', 'mean', 'std'
uns: 'X_name', 'hvg', 'log1p', 'pca', '_ot_alignment', '_supbiot'
obsm: 'X_pca', 'X_supbiot'
varm: 'PCs'
layers: 'logcounts'
from sklearn.metrics import normalized_mutual_info_score
unlabeled_category = "Unknown" # set to whatever you used
y_true = adata.obs["cell_type"]
y_pred = adata.obs["pred_cell_type"]
y_semi = adata.obs["semi_cell_type"]
# remove cells that have a semi_cell_type label (i.e., keep only unlabeled cells)
mask = (
y_true.notna()
& y_pred.notna()
& y_semi.notna()
& (y_semi.astype(str) == unlabeled_category)
)
nmi = normalized_mutual_info_score(
y_true[mask].astype(str).to_numpy(),
y_pred[mask].astype(str).to_numpy(),
average_method="arithmetic",
)
print(f"NMI (only cells with semi_cell_type == {unlabeled_category!r}) = {nmi:.6f}")
print(f"Used {mask.sum()} / {len(mask)} cells")
NMI (only cells with semi_cell_type == 'Unknown') = 0.821125
Used 6321 / 14890 cells
Evaluate#
Inspect prediction confidence and consolidate predicted labels.
ax = sc.pl.violin(adata, keys="pred_confidence", groupby="pred_cell_type", rotation=90, show=False)
ax.get_legend().remove()
plt.tight_layout()
plt.show()
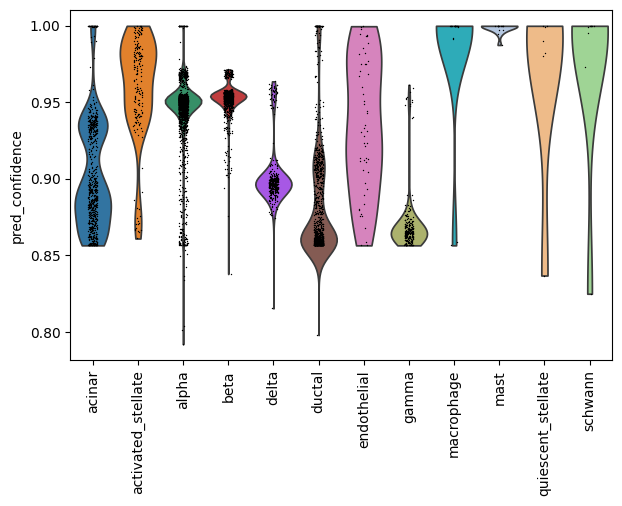
print(adata.obs["pred_cell_type"].isna().sum())
# combine labels and unlabels
adata.obs["pred_cell_type"] = (
adata.obs["pred_cell_type"].astype("string")
.fillna(adata.obs["semi_cell_type"].astype("string"))
)
print(adata.obs["pred_cell_type"].isna().sum())
8569
0
methods = ["X_supbiot"] # , "scBIOT_OT"
leiden_methods = [f'{method}_leiden' for method in methods]
for method, leiden_method in zip(methods, leiden_methods):
sc.pp.neighbors(adata, use_rep=method)
sc.tl.umap(adata)
adata.obsm[f"X_umap_{method}"] = adata.obsm["X_umap"].copy()
sc.tl.leiden(adata, key_added=leiden_method, resolution=0.8)
Visualize#
Compare batch and Leiden structure across embeddings in UMAP panels.
import matplotlib.pyplot as plt
import scanpy as sc
n_rows = 4
fig, axes = plt.subplots(
n_rows,
len(methods),
figsize=(4 * len(methods), 3.2 * n_rows),
squeeze=False
)
row_titles = ["batch", "leiden", "cell_type", "pred_cell_type"]
for col, method in enumerate(methods):
basis = f"X_umap_{method}" # or f"umap_{method}" if you store in obsm["X_umap_{method}"]
leiden_key = f"{method}_leiden"
# Row 0: batch
sc.pl.embedding(
adata,
basis=basis,
color="batch",
frameon=False,
ax=axes[0, col],
show=False,
legend_loc="on data",
legend_fontsize=10,
title=f"{method}"
)
# Row 1: leiden
sc.pl.embedding(
adata,
basis=basis,
color=leiden_key,
frameon=False,
ax=axes[1, col],
show=False,
legend_loc="on data",
legend_fontsize=10,
title=None
)
# Row 2: cell_type
sc.pl.embedding(
adata,
basis=basis,
color="cell_type",
frameon=False,
ax=axes[2, col],
show=False,
legend_loc="on data",
legend_fontsize=10,
title=None
)
# Row 3: pred_cell_type
sc.pl.embedding(
adata,
basis=basis,
color="pred_cell_type",
frameon=False,
ax=axes[3, col],
show=False,
legend_loc="on data",
legend_fontsize=10,
title=None
)
# Optional: add row labels on the left-most column
for r, lab in enumerate(row_titles):
axes[r, 0].set_ylabel(lab)
plt.tight_layout()
# fig.savefig("batch_and_annotations_per_embedding.pdf", dpi=300, bbox_inches="tight")
# plt.close(fig)
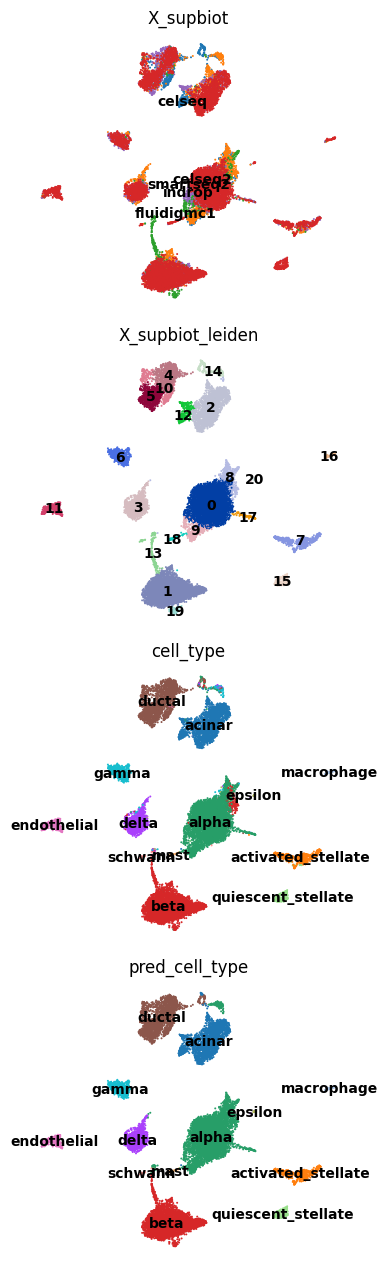
Benchmark#
fix the bug in the scib-metrics: change _graph_connectivity.py: <mask = labels == label> to <mask = (labels == label).to_numpy()>
bm = Benchmarker(
adata,
batch_key="batch",
label_key="cell_type",
bio_conservation_metrics=BioConservation(),
batch_correction_metrics=BatchCorrection(),
embedding_obsm_keys=["X_pca", "X_supbiot"],
n_jobs=-1
)
bm.benchmark()
Computing neighbors: 100%|██████████| 2/2 [00:01<00:00, 1.75it/s]
Embeddings: 100%|██████████| 2/2 [00:31<00:00, 15.86s/it]
bm.plot_results_table(min_max_scale=False)
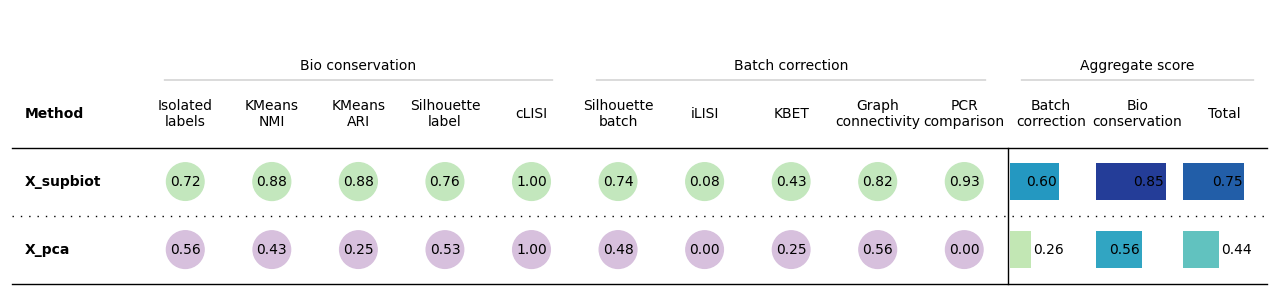
<plottable.table.Table at 0x7763a9ff63c0>