Setup#
Import dependencies, set seeds, and configure paths and plotting defaults.
import warnings
warnings.filterwarnings("ignore")
import numpy as np
import scanpy as sc
import seaborn as sns
# %pip install torch torchvision torchaudio --index-url https://download.pytorch.org/whl/cu124
import os
import pandas as pd
import scbiot as scb
from scbiot.utils import set_seed
import harmonypy as hm
from umap import UMAP
# %pip install scib-metrics
from scib_metrics.benchmark import Benchmarker, BioConservation, BatchCorrection
set_seed(42)
from pathlib import Path
dir = Path.cwd()
print(dir)
parent_dir = dir.parent
print(parent_dir)
from scimorph.theme_publication import theme_publication
from scimorph.utils import set_seed
theme_publication()
scbiot version 1.1.7
Random seed set as 42
/home/figo/software/python_libs/scbiot/examples
/home/figo/software/python_libs/scbiot
Load#
Read the paired multiome dataset from disk.
adata_path = f"{dir}/inputs/multiome.h5ad"
adata = sc.read(
adata_path,
backup_url="https://figshare.com/ndownloader/files/59742665"
)
Preprocess#
Split GEX/ATAC modalities and build PCA (RNA) plus LSI (ATAC) features.
# split to gex and peaks
gex_vars = adata.var['feature_types'] == 'GEX'
adata_gex = adata[:, gex_vars].copy()
# Filter for ATAC-related variables
atac_vars = adata.var['feature_types'] == 'ATAC'
adata_atac = adata[:, atac_vars].copy()
# 0) ATAC preprocessing (peak filtering -> LSI -> GA + smoothing)
# figshare link: https://figshare.com/ndownloader/files/59742641
gtf_file = f"{dir}/inputs/gencode.v48.chr_patch_hapl_scaff.basic.annotation.gtf.gz"
adata_ga = scb.pp.create_gene_activity(adata_atac, adata_gex, gtf_file=gtf_file, verbose=True)
adata_ga
Removed 28,633 promoter-proximal peaks (2000bp upstream / 500bp downstream). Remaining: 87,857
Running Iterative LSI iteration 1 ...
Running Iterative LSI iteration 2 ...
[GA] Kept 59,754/86,364 genes by biotype ['protein_coding', 'lncRNA']
[GA] Peaks contigs: 29; Genes contigs: 501; Common: 29
[GA] Using gene field: gene_name
[GA] Built GA with shape (69249, 35570) (cells × genes) from 116,490 peaks.
[names] Harmonized symbols; overlaps (case-insensitive): 11,874
AnnData object with n_obs × n_vars = 69249 × 35570
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'n_peaks'
uns: 'provenance'
layers: 'ga', 'ga_smooth'
import anndata
# adata_gex.write_h5ad('adata_gex.h5ad')
# adata_ga.write_h5ad('adata_ga.h5ad')
# adata_atac.write_h5ad('adata_atac.h5ad')
adata_gex = anndata.read_h5ad('adata_gex.h5ad')
adata_ga = anndata.read_h5ad('adata_ga.h5ad')
adata_atac = anndata.read_h5ad('adata_atac.h5ad')
adata_gex.obs["cell_type"] = adata_gex.obs["cell_type"]
adata_ga.obs["cell_type"] = "Unknown"
adata_gex.X = adata_gex.layers['counts']
adata = scb.pp.coembed_pca(
adata_gex, adata_ga,
label="modality",
mode='paired',
# batch_key='batch',
keys=("reference", "query"),
reference_layer="counts",
query_layer='ga_smooth',
out_key="X_shared_pca",
)
adata
AnnData object with n_obs × n_vars = 138498 × 37127
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'modality', 'obs_original'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_shared_pca'
layers: 'counts', 'rna_log1p', 'ga', 'ga_smooth', 'ga_log1p'
Integrate#
Run paired OT integration using PCA and LSI views.
adata, metrics = scb.ot.integrate(
adata,
obsm_key="X_shared_pca",
batch_key="modality",
prealign='ot',
prealign_strength=0.8,
align_reference=True,
label_key="cell_type",
unlabeled_category="Unknown",
out_key="X_supbiot"
)
======== Stage1: supervised OT for label propagation ========
[prealign] OT-Gaussian enabled target=auto strength=0.8
[baseline] KNN backend=FAISS-GPU mix=0.0227 strain=0.00000
[iter 01] mix=0.020 overlap0=0.950 strain=0.00061 floor~0.600 J=0.165 best_it=1
[iter 02] mix=0.018 overlap0=0.915 strain=0.00217 floor~0.607 J=0.160 best_it=1
[iter 03] mix=0.018 overlap0=0.913 strain=0.00219 floor~0.614 J=0.159 best_it=1
[iter 04] mix=0.018 overlap0=0.911 strain=0.00222 floor~0.621 J=0.157 best_it=1
[early stop] plateau reached.
[final] it*=1 mix=0.020 overlap0=0.950 strain=0.00061 tw=1.000
======== Stage2: Global OT for mapping query to reference ========
[align_reference] mix=0.161 overlap0=0.457 strain=0.15712 tw=0.990
Label transfer (supBIOT)#
adata = scb.ot.supbiot(
adata,
rep_key="X_supbiot",
label_key="cell_type",
unlabeled_category="Unknown",
pred_label_key='pred_cell_type',
pred_conf_key="pred_confidence",
min_conf=0.
)
adata_ga = adata[adata.obs['modality'] == 'query'].copy()
adata_gex = adata[adata.obs['modality'] == 'reference'].copy()
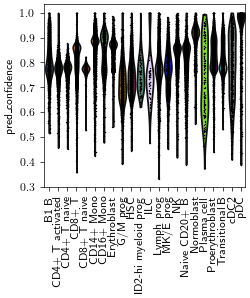
adata_ga.obs['pred_cell_type'].value_counts()
pred_cell_type
CD14+ Mono 12230
CD8+ T 11225
NK 7343
CD4+ T activated 5824
Naive CD20+ B 5746
CD4+ T naive 5660
Erythroblast 4131
Proerythroblast 3686
Lymph prog 2301
Transitional B 2145
HSC 1452
B1 B 1320
pDC 1083
Normoblast 1058
G/M prog 1043
MK/E prog 914
CD16+ Mono 881
cDC2 682
CD8+ T naive 223
Plasma cell 162
ID2-hi myeloid prog 82
ILC 58
Name: count, dtype: int64
sc.pl.violin(adata_ga, keys="pred_confidence", groupby="pred_cell_type", rotation=90)
sc.pp.neighbors(adata, use_rep="X_supbiot", n_neighbors=50, metric="cosine")
sc.tl.umap(adata, min_dist=0.3, spread=1.0, random_state=0)
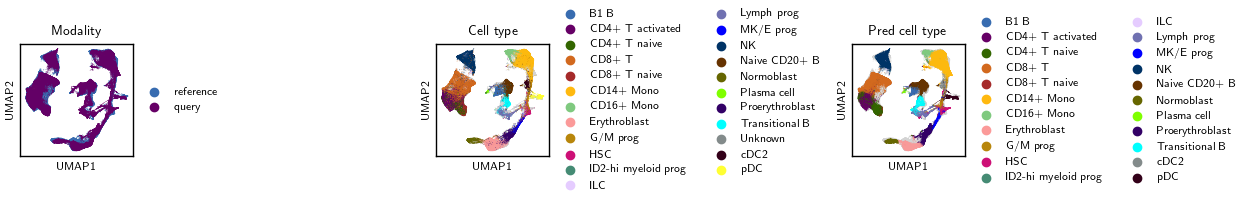
Visualize#
Compare batch and Leiden structure across embeddings in UMAP panels.
import numpy as np
import pandas as pd
import matplotlib as mpl
import matplotlib.pyplot as plt
import scanpy as sc
sc.settings._vector_friendly = True
mpl.rcParams["axes.edgecolor"] = "black"
mpl.rcParams["axes.linewidth"] = 1.0
def force_border(ax):
ax.set_axis_on()
ax.set_frame_on(True)
ax.patch.set_visible(True)
for side in ax.spines.values():
side.set_visible(True)
side.set_color("black")
side.set_linewidth(1.0)
def mask_drop_unknown_na(adata, key, drop=("Unknown", "Unkown", "NA", "NaN", "None", "")):
s = adata.obs[key].astype("string")
low = s.str.strip().str.lower()
drop_low = {d.lower() for d in drop}
m = s.notna() & (~low.isin(drop_low))
return m.to_numpy(dtype=bool, na_value=False)
methods = ["X_supbiot"]
m_cell = mask_drop_unknown_na(adata, "cell_type")
m_pred = mask_drop_unknown_na(adata, "pred_cell_type")
ncols = 3 * len(methods)
fig, axes = plt.subplots(1, ncols, figsize=(4.2 * ncols, 4.2), squeeze=False)
axes = axes[0]
for i, method in enumerate(methods):
axL, axM, axR = axes[3*i], axes[3*i + 1], axes[3*i + 2]
sc.pl.embedding(
adata, basis="umap", color="modality",
frameon=True, ax=axL, show=False,
legend_loc="right margin", legend_fontsize=8, title="Modality"
)
axL.set_box_aspect(1)
axL.set_xlabel("UMAP1"); axL.set_ylabel("UMAP2")
force_border(axL)
xlim, ylim = axL.get_xlim(), axL.get_ylim()
# hide Unknown/NA cells via mask_obs (no slicing -> no view copy issues)
sc.pl.embedding(
adata, basis="umap", color="cell_type",
mask_obs=m_cell, na_in_legend=False,
frameon=True, ax=axM, show=False,
legend_loc="right margin", legend_fontsize=8, title="Cell type"
)
axM.set_xlim(xlim); axM.set_ylim(ylim)
axM.set_box_aspect(1)
axM.set_xlabel("UMAP1"); axM.set_ylabel("UMAP2")
force_border(axM)
sc.pl.embedding(
adata, basis="umap", color="pred_cell_type",
mask_obs=m_pred, na_in_legend=False,
frameon=True, ax=axR, show=False,
legend_loc="right margin", legend_fontsize=8, title="Pred cell type"
)
axR.set_xlim(xlim); axR.set_ylim(ylim)
axR.set_box_aspect(1)
axR.set_xlabel("UMAP1"); axR.set_ylabel("UMAP2")
force_border(axR)
plt.tight_layout()
Evaluate#
import numpy as np
import pandas as pd
import scipy.sparse as sp
import anndata as ad
def collapse_paired_keep_query_pred(
adata,
emb_key="X_supbiot",
out_key="X_ot",
modality_key="modality",
ref_label="reference",
qry_label="query",
w_ref=0.5,
w_qry=0.5,
pred_type_key="pred_cell_type",
pred_conf_key_candidates=("pred_confidence", "pred_confidenc"),
*,
keep_X=True,
keep_layers=True,
keep_obsm=True,
keep_uns_colors=True,
):
# --- pull arrays ---
Xemb = np.asarray(adata.obsm[emb_key], dtype=np.float32)
mod = adata.obs[modality_key].astype(str)
# --- pairing id: strip "::reference"/"::query" ---
cell_id = pd.Index(adata.obs_names).str.replace(r"::.*$", "", regex=True)
ref_mask = (mod == ref_label).to_numpy()
qry_mask = (mod == qry_label).to_numpy()
ref_ids = pd.Index(cell_id[ref_mask])
qry_ids = pd.Index(cell_id[qry_mask])
common = ref_ids.intersection(qry_ids)
if len(common) == 0:
raise ValueError("No paired cell_ids found between reference and query.")
# positions in the original stacked adata (aligned to `common`)
ref_pos = np.flatnonzero(ref_mask)[ref_ids.get_indexer(common)].astype(np.int64, copy=False)
qry_pos = np.flatnonzero(qry_mask)[qry_ids.get_indexer(common)].astype(np.int64, copy=False)
# --- fuse embedding ---
X_fused = (w_ref * Xemb[ref_pos] + w_qry * Xemb[qry_pos]).astype(np.float32)
# --- build obs (use reference rows as carrier) ---
obs_ref = adata.obs.iloc[ref_pos].copy()
obs_ref.index = common.astype(str)
# --- build X/var ---
if keep_X:
X_ref = adata.X[ref_pos, :]
# copy to detach
X_ref = X_ref.copy() if sp.issparse(X_ref) else np.asarray(X_ref).copy()
var = adata.var.copy()
else:
X_ref = np.zeros((len(ref_pos), 0), dtype=np.float32)
var = pd.DataFrame(index=pd.Index([], name=getattr(adata.var_names, "name", None)))
adata_1n = ad.AnnData(X=X_ref, obs=obs_ref, var=var)
# --- layers (row-subset only; no obsp involvement) ---
if keep_layers:
for k, L in adata.layers.items():
try:
Ls = L[ref_pos, :]
adata_1n.layers[k] = Ls.copy() if sp.issparse(Ls) else np.asarray(Ls).copy()
except Exception:
# skip incompatible layers silently
pass
# --- obsm (row-subset only) ---
if keep_obsm:
for k, M in adata.obsm.items():
try:
if getattr(M, "shape", None) is not None and M.shape[0] == adata.n_obs:
Ms = M[ref_pos]
adata_1n.obsm[k] = Ms.copy() if hasattr(Ms, "copy") else np.asarray(Ms).copy()
except Exception:
pass
# put fused embedding
adata_1n.obsm[out_key] = X_fused
# --- copy some useful uns (colors only, avoids neighbors payloads) ---
if keep_uns_colors:
for k, v in adata.uns.items():
if isinstance(k, str) and k.endswith("_colors"):
try:
adata_1n.uns[k] = v.copy() if hasattr(v, "copy") else v
except Exception:
pass
# --- find which pred_conf column you actually have ---
pred_conf_key = None
for k in pred_conf_key_candidates:
if k in adata.obs:
pred_conf_key = k
break
# --- copy query predictions onto collapsed obs ---
q_obs = adata.obs.iloc[qry_pos].copy()
q_obs.index = common.astype(str)
if pred_type_key in q_obs.columns:
adata_1n.obs[pred_type_key] = q_obs[pred_type_key].to_numpy()
# preserve categories if categorical
if pd.api.types.is_categorical_dtype(q_obs[pred_type_key]):
adata_1n.obs[pred_type_key] = pd.Categorical(
adata_1n.obs[pred_type_key],
categories=q_obs[pred_type_key].cat.categories,
)
if pred_conf_key is not None and pred_conf_key in q_obs.columns:
adata_1n.obs[pred_conf_key] = q_obs[pred_conf_key].to_numpy()
return adata_1n
# usage
adata_1n = collapse_paired_keep_query_pred(
adata,
emb_key="X_supbiot",
out_key="X_ot",
w_ref=0.5,
w_qry=0.5,
)
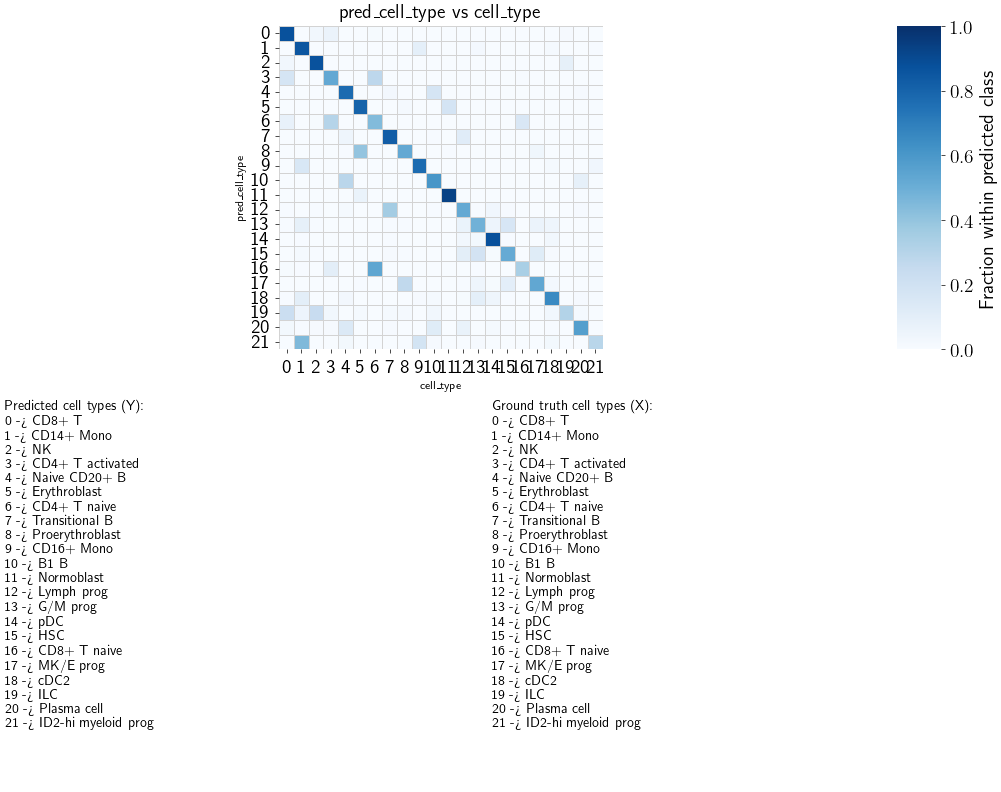
fig, ax, counts_df, norm_df = scb.pl.plot_anndata_confusion(
adata_1n,
true_key="cell_type",
pred_key="pred_cell_type",
drop_unknown=False, # key line
normalize="pred",
annotate_mapping=True,
return_data=True,
)
import numpy as np
from sklearn.neighbors import NearestNeighbors
# 1) Robustify embedding (median/MAD + clipping)
X = adata_1n.obsm["X_ot"].astype(np.float32, copy=True)
med = np.median(X, axis=0)
mad = np.median(np.abs(X - med), axis=0) + 1e-9
Z = (X - med) / (1.4826 * mad)
Z = np.clip(Z, -5, 5).astype(np.float32)
adata_1n.obsm["X_shared_pca_robust"] = Z
# 2) kNN outlier score (mean distance)
k = 30
nbrs = NearestNeighbors(n_neighbors=k).fit(Z)
d, _ = nbrs.kneighbors(Z)
knn_mean = d[:, 1:].mean(1)
# 3) Flag outliers within each reference cell type
ref_mask = adata_1n.obs["cell_type"].to_numpy() != "Unknown"
cell_type = adata_1n.obs["cell_type"].to_numpy()
outlier = np.zeros(len(adata_1n), dtype=bool)
for ct in np.unique(cell_type[ref_mask]):
idx = np.where(ref_mask & (cell_type == ct))[0]
if idx.size < 30: # don’t prune tiny groups
continue
thr = np.quantile(knn_mean[idx], 0.98) # adjust 0.98–0.995
outlier[idx] = knn_mean[idx] > thr
# 4) Relabel outlier reference cells so they’re not fixed anchors
adata_1n.obs.loc[outlier, "cell_type"] = "Unknown"
adata_1n.obs['cell_type'].value_counts()
cell_type
CD8+ T 11357
CD14+ Mono 10626
NK 6790
CD4+ T activated 5415
Naive CD20+ B 4950
Erythroblast 4817
CD4+ T naive 4310
Transitional B 2753
Proerythroblast 2254
CD16+ Mono 1856
B1 B 1852
Normoblast 1744
Lymph prog 1743
Unknown 1395
G/M prog 1178
pDC 1167
HSC 1050
CD8+ T naive 991
MK/E prog 866
cDC2 841
ILC 818
Plasma cell 371
ID2-hi myeloid prog 105
Name: count, dtype: int64
adata, metrics = scb.ot.integrate(
adata_1n,
obsm_key="X_ot",
batch_key="batch",
label_key="cell_type",
unlabeled_category="Unknown",
out_key="X_supbiot2"
)
======== Stage1: supervised OT for label propagation ========
[baseline] KNN backend=FAISS-GPU mix=1.2390 strain=0.00000
[iter 01] mix=1.359 overlap0=0.836 strain=0.00325 floor~0.600 J=0.210 best_it=1
[iter 02] mix=1.499 overlap0=0.698 strain=0.00958 floor~0.607 J=0.328 best_it=2
[iter 03] mix=1.617 overlap0=0.575 strain=0.02032 floor~0.614 J=0.415 best_it=3
[iter 04] mix=1.682 overlap0=0.504 strain=0.03680 floor~0.621 J=0.454 best_it=4
[iter 05] mix=1.736 overlap0=0.454 strain=0.05288 floor~0.629 J=0.467 best_it=5
[iter 06] mix=1.774 overlap0=0.404 strain=0.07527 floor~0.636 J=0.437 best_it=5
[iter 07] mix=1.803 overlap0=0.367 strain=0.11144 floor~0.643 J=0.373 best_it=5
[iter 08] mix=1.803 overlap0=0.369 strain=0.11322 floor~0.650 J=0.368 best_it=5
[early stop] plateau reached.
[final] it*=5 mix=1.736 overlap0=0.454 strain=0.05288 tw=0.982
======== Stage2: unsupervised OT for batch integration ========
[baseline] KNN backend=FAISS-GPU mix=1.7357 strain=0.00000
[iter 01] mix=1.752 overlap0=0.941 strain=0.00035 floor~0.600 J=0.178 best_it=1
[iter 02] mix=1.766 overlap0=0.900 strain=0.00124 floor~0.607 J=0.191 best_it=2
[iter 03] mix=1.779 overlap0=0.859 strain=0.00462 floor~0.614 J=0.192 best_it=2
[iter 04] mix=1.780 overlap0=0.859 strain=0.00477 floor~0.621 J=0.193 best_it=4
[iter 05] mix=1.794 overlap0=0.816 strain=0.00997 floor~0.629 J=0.192 best_it=4
[iter 06] mix=1.793 overlap0=0.812 strain=0.00930 floor~0.636 J=0.190 best_it=4
[iter 07] mix=1.795 overlap0=0.802 strain=0.01144 floor~0.643 J=0.183 best_it=4
[early stop] plateau reached.
[final] it*=4 mix=1.780 overlap0=0.859 strain=0.00477 tw=0.999
[label transfer] skipped; pass label_key to compute alignment metadata
======== Stage3: supervised OT for refinement ========
[baseline] KNN backend=FAISS-GPU mix=1.7803 strain=0.00000
[iter 01] mix=1.794 overlap0=0.860 strain=0.01485 floor~0.600 J=0.108 best_it=1
[iter 02] mix=1.805 overlap0=0.792 strain=0.03080 floor~0.607 J=0.122 best_it=2
[iter 03] mix=1.820 overlap0=0.707 strain=0.05641 floor~0.614 J=0.086 best_it=2
[iter 04] mix=1.829 overlap0=0.692 strain=0.06196 floor~0.621 J=0.080 best_it=2
[iter 05] mix=1.824 overlap0=0.719 strain=0.05876 floor~0.629 J=0.096 best_it=2
[early stop] plateau reached.
[final] it*=2 mix=1.805 overlap0=0.792 strain=0.03080 tw=0.999
from sklearn.metrics import normalized_mutual_info_score
y_true = adata_1n.obs["cell_type"].astype(str)
y_pred = adata_1n.obs["pred_cell_type"].astype(str)
# keep only valid rows
mask = y_true.notna() & y_pred.notna()
# (optional) ignore "unknown" labels
# mask &= (y_true != "unknown") & (y_pred != "unknown")
nmi = normalized_mutual_info_score(
y_true[mask].to_numpy(),
y_pred[mask].to_numpy(),
average_method="arithmetic",
)
print(f"NMI = {nmi:.6f}")
NMI = 0.705543
methods = ["X_supbiot2"] # , "scBIOT_OT"
leiden_methods = [f'{method}_leiden' for method in methods]
for method, leiden_method in zip(methods, leiden_methods):
sc.pp.neighbors(adata_1n, use_rep=method)
sc.tl.umap(adata_1n)
adata.obsm[f"X_umap_{method}"] = adata.obsm["X_umap"].copy()
sc.tl.leiden(adata_1n, key_added=leiden_method, resolution=0.8)
import matplotlib.pyplot as plt
import scanpy as sc
# 2 rows x len(methods) columns
fig, axes = plt.subplots(
2,
len(methods),
figsize=(4 * len(methods), 8),
squeeze=False # ensures axes is a 2D array
)
for col, method in enumerate(methods):
# 1) Top row (row=0): color by "batch"
sc.pl.embedding(
adata_1n,
basis=f"X_umap_{method}", # The coordinates stored in adata.obsm["X_umap_{method}"]
color="batch", # Assume adata.obs["batch"] exists
frameon=False,
ax=axes[0, col],
show=False,
legend_loc="on data",
legend_fontsize=10, # smaller font
title=f"{method}"
)
# 2) Bottom row (row=1): color by the Leiden clusters for this method
leiden_key = f"{method}_leiden"
sc.pl.embedding(
adata_1n,
basis=f"X_umap_{method}",
color='cell_type', # Column in adata.obs
frameon=False,
ax=axes[1, col],
show=False,
legend_loc="on data",
legend_fontsize=10, # smaller font
# title=f"{method}"
)
plt.tight_layout()
# fig.savefig("batch_and_leiden_per_embedding.pdf", dpi=300)
# plt.close(fig)

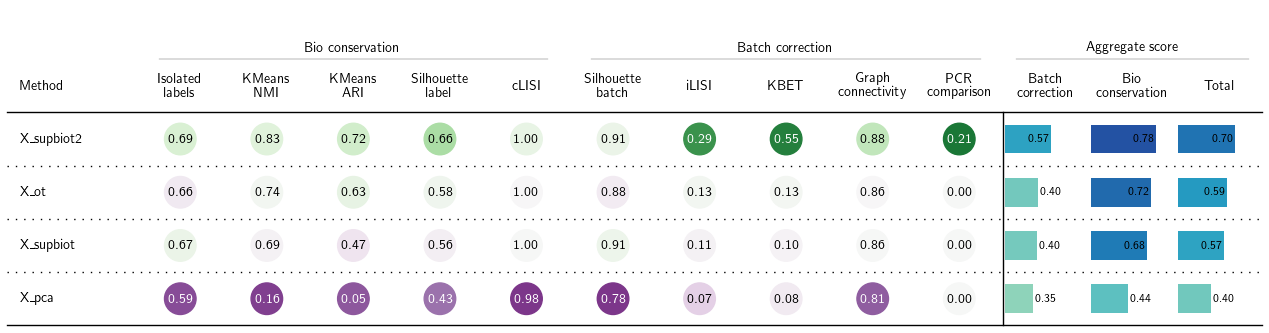
bm = Benchmarker(
adata_1n,
batch_key="batch",
label_key="cell_type",
bio_conservation_metrics=BioConservation(),
batch_correction_metrics=BatchCorrection(),
embedding_obsm_keys=["X_pca", "X_supbiot", "X_ot", "X_supbiot2"],
n_jobs=32
)
bm.benchmark()
bm.plot_results_table(min_max_scale=False)
Computing neighbors: 100%|██████████| 4/4 [00:14<00:00, 3.50s/it]
Embeddings: 100%|██████████| 4/4 [02:33<00:00, 38.27s/it]
<plottable.table.Table at 0x7d92c03d92b0>