b. label transfer from scRNA-seq to Xenium#
Define data sources and import spatial + scRNA-seq tooling for the Xenium workflow.
# https://www.10xgenomics.com/datasets/xenium-prime-ffpe-human-ovarian-cancer
# https://squidpy.readthedocs.io/en/stable/notebooks/tutorials/tutorial_xenium.html
# https://colab.research.google.com/github/10XGenomics/analysis_guides/blob/main/Xenium_5k_data_analysis_journey_python.ipynb#scrollTo=ca7d0356
from __future__ import annotations
import os
from pathlib import Path
import numpy as np
import pandas as pd
import scanpy as scm
import squidpy as sq
import spatialdata as sd
from spatialdata_io import xenium
import scbiot as scb
import scanpy as sc
import matplotlib.pyplot as plt
import seaborn as sns
import matplotlib as mpl
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/_utils/__init__.py:33: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
from anndata import __version__ as anndata_version
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/__init__.py:24: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
if Version(anndata.__version__) >= Version("0.11.0rc2"):
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/readwrite.py:16: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
if Version(anndata.__version__) >= Version("0.11.0rc2"):
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/dask/dataframe/__init__.py:31: FutureWarning: The legacy Dask DataFrame implementation is deprecated and will be removed in a future version. Set the configuration option `dataframe.query-planning` to `True` or None to enable the new Dask Dataframe implementation and silence this warning.
warnings.warn(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/xarray_schema/__init__.py:1: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import DistributionNotFound, get_distribution
scbiot version 1.1.7
ROOT = Path(os.environ.get("SCBIOT_EXAMPLES_PATH", Path.cwd()))
print(ROOT)
DATA_DIR = ROOT / "inputs" / "xenium"
xenium_dir = DATA_DIR / "Xenium_Prime_Ovarian_Cancer_FFPE_XRrun_outs"
zarr_path = ROOT / "inputs" / "xenium" / "Xenium_Prime_Ovarian_Cancer.zarr"
rna_h5 = DATA_DIR / "reference" / "17k_Ovarian_Cancer_scFFPE_count_filtered_feature_bc_matrix.h5"
cell_type = DATA_DIR / "reference" / "FLEX_Ovarian_Barcode_Cluster_Annotation.csv"
# sdata = xenium(xenium_dir,
# transcripts=False, # <-- key
# cells_as_circles=True # to silence the deprecation warning)
# )
# # Convert to Zarr.
# sdata.write(zarr_path, overwrite=True)
/home/figo/software/python_libs/scbiot/examples
rna_h5
PosixPath('/home/figo/software/python_libs/scbiot/examples/inputs/xenium/reference/17k_Ovarian_Cancer_scFFPE_count_filtered_feature_bc_matrix.h5')
Load Xenium spatial data#
Open the Xenium SpatialData Zarr and extract the table as AnnData.
sdata = sd.read_zarr(zarr_path)
sdata
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/zarr/creation.py:610: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/zarr/creation.py:610: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/zarr/creation.py:610: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/zarr/creation.py:610: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/zarr/creation.py:610: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/zarr/creation.py:610: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
SpatialData object, with associated Zarr store: /home/figo/software/python_libs/scbiot/examples/inputs/xenium/Xenium_Prime_Ovarian_Cancer.zarr
├── Images
│ └── 'morphology_focus': DataTree[cyx] (4, 37631, 54089), (4, 18815, 27044), (4, 9407, 13522), (4, 4703, 6761), (4, 2351, 3380)
├── Labels
│ ├── 'cell_labels': DataTree[yx] (37631, 54089), (18815, 27044), (9407, 13522), (4703, 6761), (2351, 3380)
│ └── 'nucleus_labels': DataTree[yx] (37631, 54089), (18815, 27044), (9407, 13522), (4703, 6761), (2351, 3380)
├── Shapes
│ ├── 'cell_boundaries': GeoDataFrame shape: (407124, 1) (2D shapes)
│ ├── 'cell_circles': GeoDataFrame shape: (407124, 2) (2D shapes)
│ └── 'nucleus_boundaries': GeoDataFrame shape: (401404, 1) (2D shapes)
└── Tables
└── 'table': AnnData (407124, 5101)
with coordinate systems:
▸ 'global', with elements:
morphology_focus (Images), cell_labels (Labels), nucleus_labels (Labels), cell_boundaries (Shapes), cell_circles (Shapes), nucleus_boundaries (Shapes)
# For the analysis we use the anndata.AnnData object
adata_query = sdata.tables["table"]
adata_query
AnnData object with n_obs × n_vars = 407124 × 5101
obs: 'cell_id', 'transcript_counts', 'control_probe_counts', 'genomic_control_counts', 'control_codeword_counts', 'unassigned_codeword_counts', 'deprecated_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'nucleus_count', 'segmentation_method', 'region', 'z_level', 'cell_labels'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'spatialdata_attrs'
obsm: 'spatial'
Preprocess#
Compute Xenium QC metrics, visualize distributions, and filter low-quality cells/genes.
sc.pp.calculate_qc_metrics(adata_query, percent_top=(10, 20, 50, 150), inplace=True)
### The percentage of control probes and control codewords can be calculated from adata.obs
cprobes = (
adata_query.obs["control_probe_counts"].sum() / adata_query.obs["total_counts"].sum() * 100
)
cwords = (
adata_query.obs["control_codeword_counts"].sum() / adata_query.obs["total_counts"].sum() * 100
)
print(f"Negative DNA probe count % : {cprobes}")
print(f"Negative decoding count % : {cwords}")
Negative DNA probe count % : 0.00075974271899767
Negative decoding count % : 0.0003784626894388888
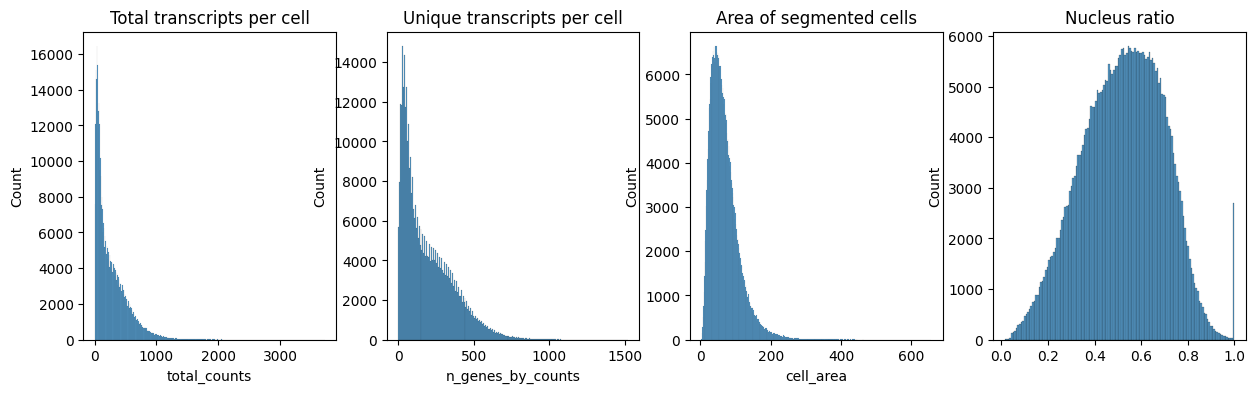
### Next we plot the distribution of total transcripts per cell, unique transcripts per cell, area of segmented cells and the ratio of nuclei area to their cells
fig, axs = plt.subplots(1, 4, figsize=(15, 4))
axs[0].set_title("Total transcripts per cell")
sns.histplot(
adata_query.obs["total_counts"],
kde=False,
ax=axs[0],
)
axs[1].set_title("Unique transcripts per cell")
sns.histplot(
adata_query.obs["n_genes_by_counts"],
kde=False,
ax=axs[1],
)
axs[2].set_title("Area of segmented cells")
sns.histplot(
adata_query.obs["cell_area"],
kde=False,
ax=axs[2],
)
axs[3].set_title("Nucleus ratio")
sns.histplot(
adata_query.obs["nucleus_area"] / adata_query.obs["cell_area"],
kde=False,
ax=axs[3],
)
<Axes: title={'center': 'Nucleus ratio'}, ylabel='Count'>
adata_query.layers["counts"] = adata_query.X.copy()
sc.pp.filter_cells(adata_query, min_counts=20)
thres = np.quantile(adata_query.obs["total_counts"], 0.98)
print(thres)
sc.pp.filter_cells(adata_query, max_counts=thres)
# # We also filter out genes that are rarely expressed
sc.pp.filter_genes(adata_query, min_cells=100)
975.0
Load scRNA-seq reference#
Read the 10x reference and map barcode-based cell types.
ref = sc.read_10x_h5(rna_h5)
print(ref)
df_celltype = pd.read_csv(cell_type)
print(df_celltype.head())
barcode_col = "Barcode"
celltype_col = "Cell Annotation"
# -----------------------------------------------
ref_barcodes = ref.obs_names.astype(str)
csv_barcodes = df_celltype[barcode_col].astype(str)
# build a mapping barcode -> cell_type
mapping = (
df_celltype.assign(_bc=csv_barcodes)[["_bc", celltype_col]]
.drop_duplicates("_bc")
.set_index("_bc")[celltype_col]
)
# assign into ref.obs
ref.obs["cell_type"] = pd.Series(ref_barcodes, index=ref.obs_names).map(mapping)
# remove NA
# remove cells with missing cell_type
adata_ref = ref[ref.obs["cell_type"].notna()].copy()
# quick sanity check
print(ref.obs["cell_type"].value_counts(dropna=False).head(20))
print("Missing cell_type:", ref.obs["cell_type"].isna().sum(), "of", ref.n_obs)
AnnData object with n_obs × n_vars = 17553 × 18082
var: 'gene_ids', 'feature_types', 'genome'
Barcode Cell Annotation
0 AAACAAGCAAAGGTAAACTTTAGG-1 Tumor Associated Fibroblasts
1 AAACAAGCAAATCACGACTTTAGG-1 Endothelial Cells
2 AAACAAGCAACTTGTCACTTTAGG-1 Stromal Associated Fibroblasts
3 AAACAAGCAAGGCCTGACTTTAGG-1 Tumor Associated Fibroblasts
4 AAACAAGCACATTCGTACTTTAGG-1 Tumor Associated Fibroblasts
cell_type
Macrophages 2327
Tumor Cells 2255
Tumor Associated Fibroblasts 2251
Proliferative Tumor Cells 1950
Stromal Associated Fibroblasts 1375
Endothelial Cells 1353
VEGFA+ Tumor Cells 1325
Smooth Muscle Cells 959
Pericytes 881
T & NK Cells 812
MT-High, Jun+/Fos+ Tumor Cells 606
NaN 503
Fallopian Tube Epithelium 298
Granulosa Cells 256
Ciliated Epithelial Cells 169
Inflammatory Tumor Cells 135
Malignant Cells Lining Cyst 98
Name: count, dtype: int64
Missing cell_type: 503 of 17553
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/anndata/_core/anndata.py:1798: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/anndata/_core/anndata.py:1798: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/anndata/_core/anndata.py:1798: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Subset for a quick speed test#
You can run full dataset by skipping this step.
# frac = 0.2
# rng = np.random.default_rng(0)
# n = int(np.floor(frac * adata_query.n_obs))
# idx = rng.choice(adata_query.n_obs, size=n, replace=False)
# adata_query = adata_query[idx, :].copy()
# adata_query
Preprocess#
Fix duplicated gene symbols and harmonize features before integration.
# Fix duplicated gene symbols
adata_query.layers['counts'] = adata_query.X.copy()
adata_ref.layers['counts'] = adata_ref.X.copy()
adata_ref.var["gene_symbol"] = adata_ref.var_names # make it a var column
adata_ref = sc.get.aggregate(
adata_ref,
by="gene_symbol",
axis="var", # aggregate features
func="sum",
layer="counts",
)
adata_ref.layers['counts'] = adata_ref.layers['sum']
adata_ref.X = adata_ref.layers['sum']
adata_ref
AnnData object with n_obs × n_vars = 17050 × 18079
obs: 'cell_type'
var: 'gene_symbol'
layers: 'sum', 'counts'
Build the reference UMAP#
Build a reference UMAP (only if you plan to reuse it) so you can map new cells onto the same coordinates when running supbiot with use_embedding_ref=True.
import numpy as np
import scipy.sparse as sp
import scanpy as sc
import anndata
import scbiot as scb
# Strore raw counts
adata_ref.layers["counts"] = adata_ref.X.copy()
adata_query.layers["counts"] = adata_query.X.copy()
adata_ref_umap = adata_ref.copy()
sc.pp.highly_variable_genes(adata_ref_umap, n_top_genes=2000, flavor="seurat_v3")
sc.pp.normalize_total(adata_ref_umap)
sc.pp.log1p(adata_ref_umap)
sc.pp.scale(adata_ref_umap)
sc.tl.pca(adata_ref_umap, n_comps=50, use_highly_variable=True)
sc.pp.neighbors(adata_ref_umap, use_rep="X_pca")
sc.tl.umap(adata_ref_umap)
# sc.pl.umap(adata_ref_umap)
# Generate umap of reference if using reference umap
adata_ref.obsm["X_pca"] = adata_ref_umap.obsm["X_pca"].copy()
adata_ref.obsm["X_umap"] = adata_ref_umap.obsm["X_umap"].copy()
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/preprocessing/_pca/__init__.py:227: FutureWarning: Argument `use_highly_variable` is deprecated, consider using the mask argument. Use_highly_variable=True can be called through mask_var="highly_variable". Use_highly_variable=False can be called through mask_var=None
mask_var_param, mask_var = _handle_mask_var(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/preprocessing/_pca/__init__.py:245: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
Version(ad.__version__) < Version("0.9")
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/neighbors/__init__.py:430: FutureWarning: Use obsm (e.g. `k in adata.obsm` or `adata.obsm.keys() | {'u'}`) instead of AnnData.obsm_keys, AnnData.obsm_keys is deprecated and will be removed in the future.
if "X_diffmap" in adata.obsm_keys():
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Concatenate adata#
Concatenate adata_ref and adata_query together
adata_ref.obs["cell_type"] = adata_ref.obs["cell_type"]
adata_query.obs["cell_type"] = "Unknown"
adata = anndata.concat([adata_ref, adata_query], join='inner', label="batch", keys=["reference", "query"])
print(adata)
# Key:store raw counts back
adata.X = adata.layers["counts"].copy()
# print(adata)
counts = adata.layers["counts"].copy()
# add umap to concated adata
# rebuild X_umap on the concatenated adata (ref only)
umap = np.full((adata.n_obs, adata_ref_umap.obsm["X_umap"].shape[1]), np.nan, dtype=float)
ref_mask = (adata.obs["batch"].values == "reference")
umap[ref_mask] = adata_ref_umap.obsm["X_umap"]
adata.obsm["X_umap"] = umap
AnnData object with n_obs × n_vars = 393657 × 4902
obs: 'cell_type', 'batch'
layers: 'counts'
PCA#
Build a shared PCA space
sc.pp.highly_variable_genes(adata, n_top_genes=2000, flavor="seurat_v3")
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.scale(adata)
sc.tl.pca(adata, n_comps=50, use_highly_variable=True)
/home/figo/.local/share/uv/python/cpython-3.12.8-linux-x86_64-gnu/lib/python3.12/functools.py:909: UserWarning: zero-centering a sparse array/matrix densifies it.
return dispatch(args[0].__class__)(*args, **kw)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/preprocessing/_pca/__init__.py:227: FutureWarning: Argument `use_highly_variable` is deprecated, consider using the mask argument. Use_highly_variable=True can be called through mask_var="highly_variable". Use_highly_variable=False can be called through mask_var=None
mask_var_param, mask_var = _handle_mask_var(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/preprocessing/_pca/__init__.py:245: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
Version(ad.__version__) < Version("0.9")
Integrate disjoint datasets with label transfer (reference scRNA-seq –> Xenium)#
Use supervised OT when integrating disjoint modalities where the reference scRNA-seq dataset is distinct from the Xenium dataset(s). In this setting, enable CORAL feature-space prealignment to reduce global distribution mismatch before running supervised OT:
prealign=”coral”
prealign_strength=1.0
This CORAL step is applied prior to supervised OT integration to improve cross-dataset alignment when there is limited overlap between datasets.
Speed: Set approximate=True to accelerate the run time.
Tradeoff: This uses a less strictly converged OT plan—often very close in practice, but it can slightly change the resulting embedding and/or label transfer
adata, metrics = scb.ot.integrate(
adata,
prealign='coral',
prealign_strength=1.,
obsm_key='X_pca',
batch_key="batch",
out_key="X_supbiot",
label_key="cell_type",
unlabeled_category="Unknown"
)
======== Stage1: supervised OT for label propagation ========
[prealign] CORAL enabled target=auto strength=1.0
[baseline] KNN backend=FAISS-GPU mix=0.1267 strain=0.00000
[iter 01] mix=0.146 overlap0=0.969 strain=0.00011 floor~0.600 J=0.199 best_it=1
[iter 02] mix=0.163 overlap0=0.948 strain=0.00035 floor~0.607 J=0.216 best_it=2
[iter 03] mix=0.178 overlap0=0.927 strain=0.00068 floor~0.614 J=0.227 best_it=3
[iter 04] mix=0.189 overlap0=0.910 strain=0.00109 floor~0.621 J=0.236 best_it=4
[iter 05] mix=0.198 overlap0=0.887 strain=0.00159 floor~0.629 J=0.237 best_it=5
[iter 06] mix=0.205 overlap0=0.873 strain=0.00220 floor~0.636 J=0.244 best_it=6
[iter 07] mix=0.209 overlap0=0.854 strain=0.00292 floor~0.643 J=0.241 best_it=6
[iter 08] mix=0.209 overlap0=0.850 strain=0.00293 floor~0.650 J=0.239 best_it=6
[iter 09] mix=0.209 overlap0=0.851 strain=0.00295 floor~0.657 J=0.240 best_it=6
[early stop] plateau reached.
[final] it*=6 mix=0.205 overlap0=0.873 strain=0.00220 tw=0.999
======== Stage2: unsupervised OT for batch integration ========
[baseline] KNN backend=FAISS-GPU mix=0.2049 strain=0.00000
[iter 01] mix=0.210 overlap0=0.875 strain=0.00990 floor~0.600 J=0.114 best_it=1
[iter 02] mix=0.214 overlap0=0.809 strain=0.03414 floor~0.607 J=0.107 best_it=1
[iter 03] mix=0.217 overlap0=0.809 strain=0.03288 floor~0.614 J=0.111 best_it=1
[iter 04] mix=0.217 overlap0=0.813 strain=0.03389 floor~0.621 J=0.112 best_it=1
[early stop] plateau reached.
[final] it*=1 mix=0.210 overlap0=0.875 strain=0.00990 tw=1.000
[label transfer] skipped; pass label_key to compute alignment metadata
======== Stage3: supervised OT for refinement ========
[baseline] KNN backend=FAISS-GPU mix=0.2102 strain=0.00000
[iter 01] mix=0.215 overlap0=0.968 strain=0.00013 floor~0.600 J=0.184 best_it=1
[iter 02] mix=0.216 overlap0=0.946 strain=0.00045 floor~0.607 J=0.184 best_it=1
[iter 03] mix=0.216 overlap0=0.943 strain=0.00047 floor~0.614 J=0.183 best_it=1
[iter 04] mix=0.216 overlap0=0.944 strain=0.00048 floor~0.621 J=0.183 best_it=1
[early stop] plateau reached.
[final] it*=1 mix=0.215 overlap0=0.968 strain=0.00013 tw=1.000
adata
AnnData object with n_obs × n_vars = 393657 × 4902
obs: 'cell_type', 'batch'
var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'mean', 'std'
uns: 'hvg', 'log1p', 'pca', '_ot_alignment', '_supbiot'
obsm: 'X_umap', 'X_pca', 'X_supbiot'
varm: 'PCs'
layers: 'counts'
adata = scb.ot.supbiot(
adata,
label_key="cell_type",
unlabeled_category="Unknown",
pred_label_key='pred_cell_type',
pred_conf_key="pred_confidence",
min_conf=0.25
)
# adata.layers["counts"] = counts
adata_query = adata[adata.obs['batch'] == 'query'].copy()
adata_ref = adata[adata.obs['batch'] == 'reference'].copy()
adata_query.obs['pred_cell_type'].value_counts()
pred_cell_type
Tumor Cells 65689
Tumor Associated Fibroblasts 44755
VEGFA+ Tumor Cells 39203
Proliferative Tumor Cells 38319
Smooth Muscle Cells 35548
Macrophages 33687
MT-High, Jun+/Fos+ Tumor Cells 27160
Stromal Associated Fibroblasts 25707
Pericytes 22137
Endothelial Cells 20581
T & NK Cells 10798
Fallopian Tube Epithelium 4161
Ciliated Epithelial Cells 2533
Granulosa Cells 2407
Malignant Cells Lining Cyst 2311
Inflammatory Tumor Cells 1611
Name: count, dtype: int64
adata_query
AnnData object with n_obs × n_vars = 376607 × 4902
obs: 'cell_type', 'batch', 'pred_cell_type', 'pred_confidence'
var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'mean', 'std'
uns: 'hvg', 'log1p', 'pca', '_ot_alignment', '_supbiot'
obsm: 'X_umap', 'X_pca', 'X_supbiot'
varm: 'PCs'
layers: 'counts'
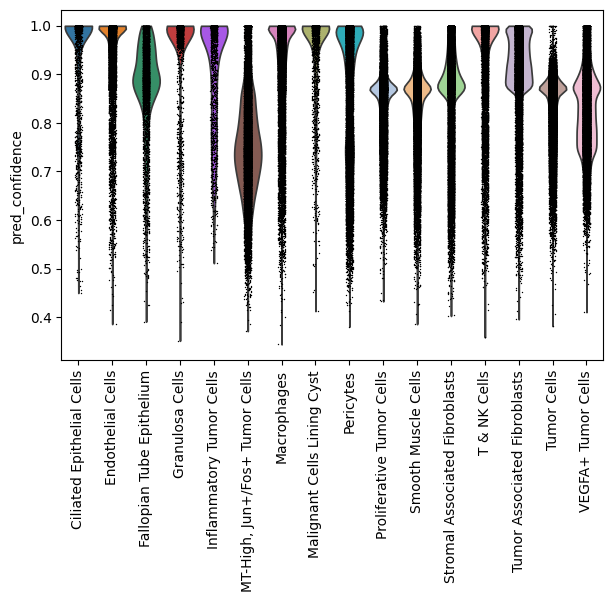
Evaluate#
Inspect predicted label confidence for query cells.
sc.pl.violin(adata_query, keys="pred_confidence", groupby="pred_cell_type", rotation=90)
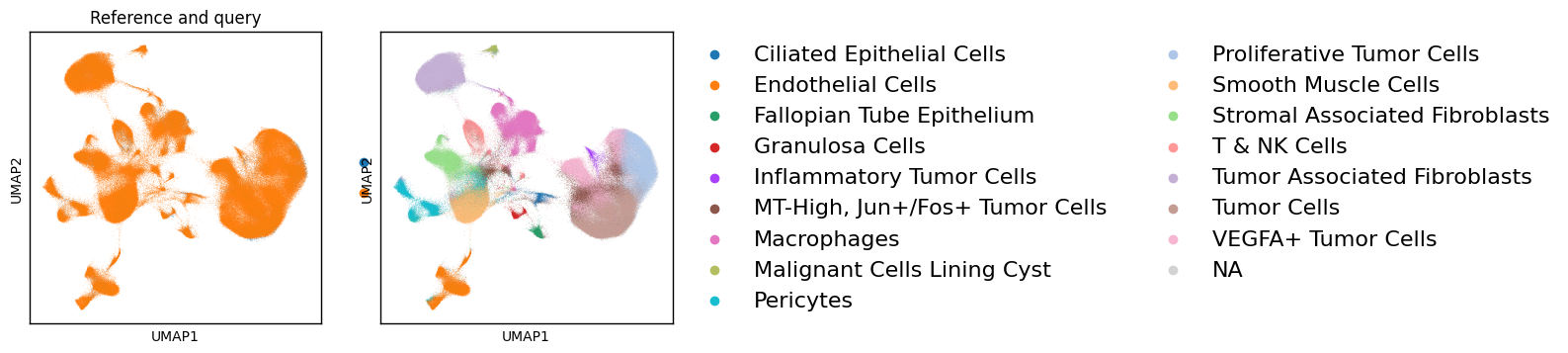
Visualize#
Compute a UMAP on the supBIOT embedding and plot batch/predicted labels.
sc.pp.neighbors(adata, use_rep='X_supbiot', n_neighbors=50, metric="cosine")
sc.tl.umap(adata, min_dist=0.3, spread=1.0, random_state=0)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/neighbors/__init__.py:430: FutureWarning: Use obsm (e.g. `k in adata.obsm` or `adata.obsm.keys() | {'u'}`) instead of AnnData.obsm_keys, AnnData.obsm_keys is deprecated and will be removed in the future.
if "X_diffmap" in adata.obsm_keys():
counts = adata_ref.obs['cell_type'].value_counts()
celltype_order = counts.index.tolist()
adata_ref.obs['cell_type'] = adata_ref.obs['cell_type'].astype(
pd.CategoricalDtype(categories=celltype_order, ordered=True) # from adata_gex
)
sc.settings._vector_friendly = True
# Make sure the default edge isn’t white or hairline
mpl.rcParams['axes.edgecolor'] = 'black'
mpl.rcParams['axes.linewidth'] = 1.0
def force_border(ax):
ax.set_axis_on() # ensure axes are on
ax.set_frame_on(True) # ensure frame is drawn
ax.patch.set_visible(True) # ensure background patch exists
for side in ax.spines.values():
side.set_visible(True)
side.set_color('black')
side.set_linewidth(1.0)
methods = ["X_supbiot"]
ncols = 2 * len(methods)
fig, axes = plt.subplots(1, ncols, figsize=(4.2*ncols, 4.2), squeeze=False)
axes = axes[0]
for i, method in enumerate(methods):
basis = f"X_umap_{method}"
axL, axR = axes[2*i], axes[2*i+1]
sc.pl.embedding(
adata, basis="X_umap", color="batch",
frameon=True, ax=axL, show=False,
legend_loc="right margin", legend_fontsize=16, title='Reference and query'
)
axL.set_box_aspect(1)
axL.set_xlabel("UMAP1"); axL.set_ylabel("UMAP2")
force_border(axL)
sc.pl.embedding(
adata, basis="X_umap", color="pred_cell_type", # "scBIOT_celltype", #"cell_type",
frameon=True, ax=axR, show=False,
legend_loc="right margin", legend_fontsize=16, title=""
)
axR.set_box_aspect(1)
axR.set_xlabel("UMAP1"); axR.set_ylabel("UMAP2")
force_border(axR)
plt.tight_layout()
# fig.savefig(f"{cwd}_batch_and_leiden_per_embedding.pdf", dpi=300, transparent=True)
/tmp/ipykernel_2506140/1606730757.py:50: UserWarning: Tight layout not applied. The left and right margins cannot be made large enough to accommodate all Axes decorations.
plt.tight_layout()
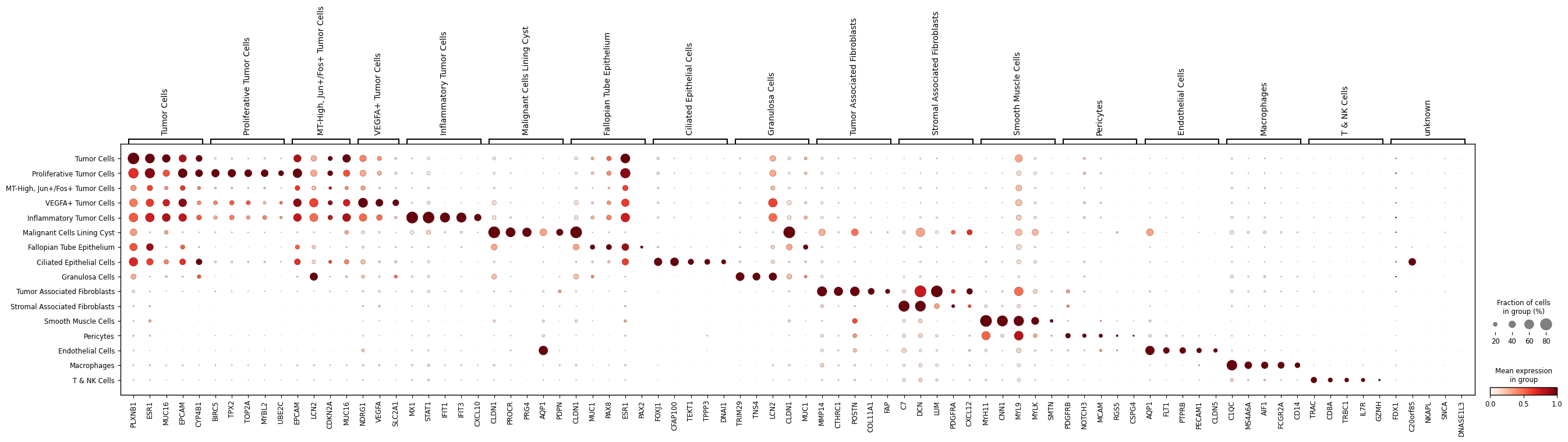
Visualize (markers)#
Plot marker gene dotplots by predicted cell type.
# -----------------------------
order = [
"Tumor Cells",
"Proliferative Tumor Cells",
"MT-High, Jun+/Fos+ Tumor Cells",
"VEGFA+ Tumor Cells",
"Inflammatory Tumor Cells",
"Malignant Cells Lining Cyst",
"Fallopian Tube Epithelium",
"Ciliated Epithelial Cells",
"Granulosa Cells",
"Tumor Associated Fibroblasts",
"Stromal Associated Fibroblasts",
"Smooth Muscle Cells",
"Pericytes",
"Endothelial Cells",
"Macrophages",
"T & NK Cells",
"unknown",
]
adata_query.obs["pred_cell_type"] = adata_query.obs["pred_cell_type"].astype("category")
# keep only categories that exist in your data (avoid errors)
order = [ct for ct in order if ct in adata_query.obs["pred_cell_type"].cat.categories]
adata_query.obs["pred_cell_type"] = adata_query.obs["pred_cell_type"].cat.reorder_categories(order, ordered=True)
# -----------------------------
# 2) Marker genes (EDIT ME)
# -----------------------------
# -----------------------------
# 2) Marker genes (UPDATED: <= 5 genes each, from your new DEGs)
# -----------------------------
markers = {
"Tumor Cells": [
"PLXNB1", "ESR1", "MUC16", "EPCAM", "CYP4B1"
],
"Proliferative Tumor Cells": [
"BIRC5", "TPX2", "TOP2A", "MYBL2", "UBE2C"
],
"MT-High, Jun+/Fos+ Tumor Cells": [
"H19", "EPCAM", "LCN2", "CDKN2A", "MUC16"
],
"VEGFA+ Tumor Cells": [
"H19", "NDRG1", "VEGFA", "LDHA", "SLC2A1"
],
"Inflammatory Tumor Cells": [
"MX1", "STAT1", "IFIT1", "IFIT3", "CXCL10"
],
"Malignant Cells Lining Cyst": [
"CLDN1", "PROCR", "PRG4", "AQP1", "PDPN"
],
"Fallopian Tube Epithelium": [
"CLDN1", "MUC1", "PAX8", "ESR1", "PAX2"
],
"Ciliated Epithelial Cells": [
"FOXJ1", "CFAP100", "TEKT1", "TPPP3", "DNAI1"
],
"Granulosa Cells": [
"TRIM29", "TNS4", "LCN2", "CLDN1", "MUC1"
],
"Tumor Associated Fibroblasts": [
"MMP14", "CTHRC1", "POSTN", "COL11A1", "FAP"
],
"Stromal Associated Fibroblasts": [
"C7", "DCN", "LUM", "PDGFRA", "CXCL12"
],
"Smooth Muscle Cells": [
"MYH11", "CNN1", "MYL9", "MYLK", "SMTN"
],
"Pericytes": [
"PDGFRB", "NOTCH3", "MCAM", "RGS5", "CSPG4"
],
"Endothelial Cells": [
"AQP1", "FLT1", "PTPRB", "PECAM1", "CLDN5"
],
"Macrophages": [
"C1QC", "MS4A6A", "AIF1", "FCGR2A", "CD14"
],
"T & NK Cells": [
"TRAC", "CD8A", "TRBC1", "IL7R", "GZMH"
],
"unknown": [
"FDX1", "C20orf85", "NKAPL", "SNCA", "DNASE1L3"
],
}
# -----------------------------
# 3) Keep only genes that exist in the object (prevents KeyErrors)
# -----------------------------
present = set(adata_query.var_names)
markers_present = {k: [g for g in v if g in present] for k, v in markers.items()}
# optionally drop empty entries (if none of their markers are present)
markers_present = {k: v for k, v in markers_present.items() if len(v) > 0}
# -----------------------------
# 4) Dotplot
sc.pl.dotplot(
adata_query,
var_names=markers_present,
groupby="pred_cell_type",
standard_scale="var",
dot_min=0.0,
dot_max=0.8,
show=True,
)
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/plotting/_anndata.py:2074: FutureWarning: Use obs (e.g. `k in adata.obs` or `str(adata.obs.columns.tolist())`) instead of AnnData.obs_keys, AnnData.obs_keys is deprecated and will be removed in the future.
if group not in [*adata.obs_keys(), adata.obs.index.name]:
Evaluate (DE)#
Rank marker genes for predicted cell types and review top hits.
sc.tl.rank_genes_groups(
adata_query,
groupby="pred_cell_type",
method="wilcoxon", # robust default
pts=True, # adds fraction-expressing metrics (pct)
use_raw=False # set True only if adata.raw holds log1p normalized data
)
# Quick visual: top markers per group (ranked genes)
sc.pl.rank_genes_groups(adata_query, n_genes=20, sharey=False)
# ----------------------------
# (A) Get a tidy dataframe for ALL groups (long format)
df_all = sc.get.rank_genes_groups_df(adata_query, group=None)
# Optional filtering: keep "useful" hits (tune thresholds)
df_filt = df_all.query("pvals_adj < 0.05 and logfoldchanges > 0.25").copy()
# Save to disk (edit path)
# df_all.to_csv("DE_cell_type_vs_rest_all.csv", index=False)
# df_filt.to_csv("DE_cell_type_vs_rest_filtered.csv", index=False)
# (B) Save one file per cell type (top 200 by adjusted p-value)
# out = {}
# for ct in adata.obs["pred_cell_type"].cat.categories if pd.api.types.is_categorical_dtype(adata.obs["pred_cell_type"]) else adata.obs["cell_type"].unique():
# d = df_all[df_all["group"] == ct].copy()
# d = d.sort_values(["scores", "pvals_adj"]).head(200)
# out[ct] = d
# safe = str(ct).replace("/", "_").replace(" ", "_")
# d.to_csv(f"DE_{safe}_vs_rest_top200.csv", index=False)
# print("Done. Example head:\n", df_filt.head())
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/tools/_rank_genes_groups.py:482: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, "logfoldchanges"] = np.log2(
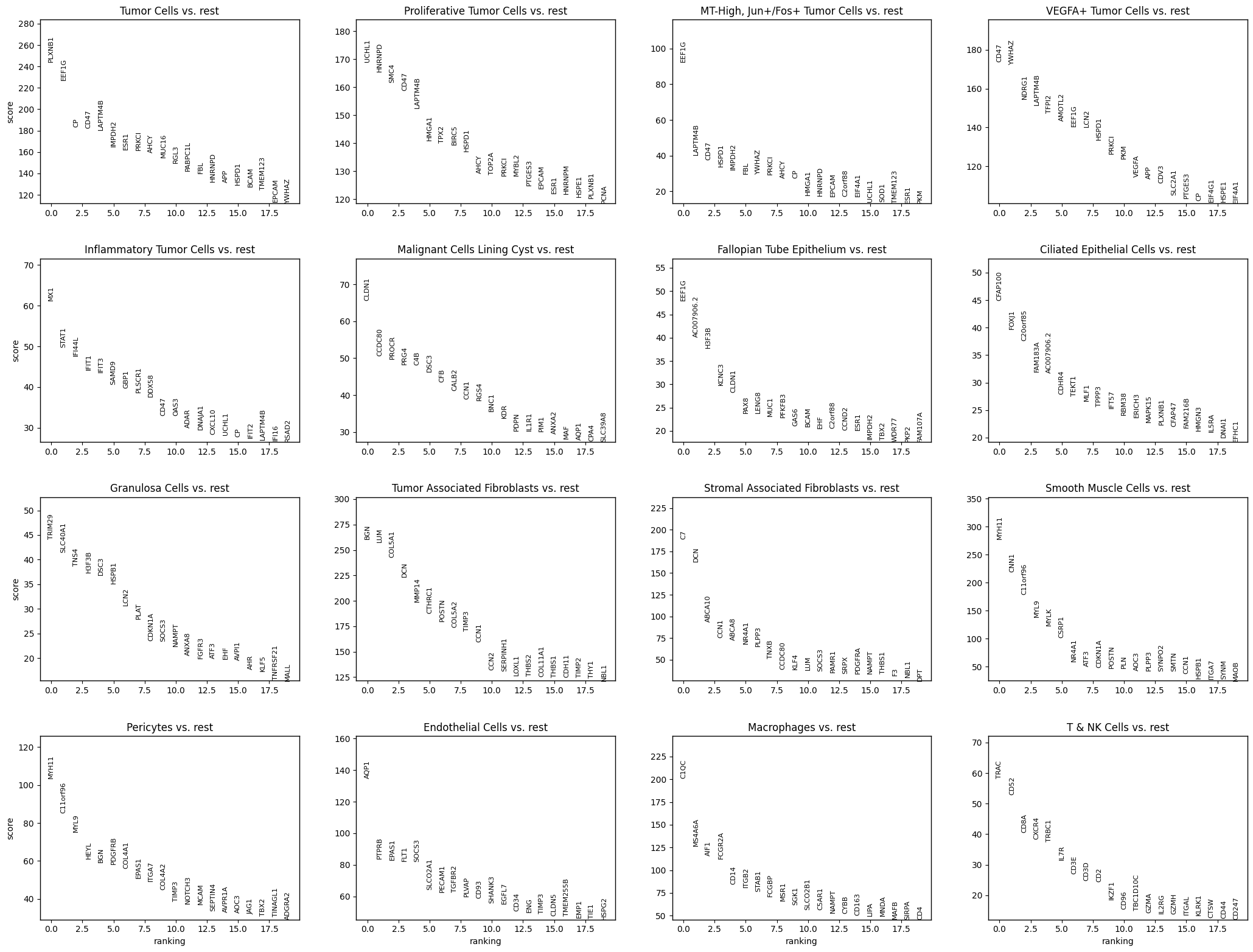
df_all = df_all.sort_values(["group", "scores"], ascending=[True, False])
for g in df_all["group"].unique():
df_g = df_all[df_all["group"] == g].head(100)
# Print as one-line list of genes
genes = df_g["names"].tolist()
print("\n" + "="*80)
print(f"pred_cell_type = {g} | Top100 genes")
print(", ".join(genes))
# Optional: if you prefer a mini table, uncomment:
# print(df_g[["names", "logfoldchanges", "pvals_adj", "scores", "pct_nz_group", "pct_nz_reference"]].head(15).to_string(index=False))
# ----------------------------
# 3) (Optional) export top100 per cluster to a single CSV
# ----------------------------
top100 = df_all.groupby("group", group_keys=False).head(100)
# top100.to_csv(f"top100_DE_by_{GROUPBY}.csv", index=False)
# print(f"\nSaved: top100_DE_by_{GROUPBY}.csv")
# top100
================================================================================
pred_cell_type = Ciliated Epithelial Cells | Top100 genes
CFAP100, FOXJ1, C20orf85, FAM183A, AC007906.2, CDHR4, TEKT1, MLF1, TPPP3, IFT57, RBM38, ERICH3, MAPK15, PLXNB1, CFAP47, FAM216B, HMGN3, IL5RA, DNAI1, EFHC1, LDLRAD1, CFAP43, NEK11, PTGES3, CYP4B1, SPAG8, DRC3, F11R, FHAD1, DZIP1L, MDM2, DNAH11, RUVBL1, SPAG17, SERPINI2, DNAH7, ODF2L, JPT2, SQLE, MAPK10, DLEC1, ZMYND10, SPATA18, DRC1, ALCAM, HIPK3, CCDC50, DNAH10, MYCBP, RFX2, DNAJC10, P4HTM, UCHL1, DNAH9, CCDC113, DNAH12, CSPP1, MYB, EPCAM, UFC1, GCLM, PTPRF, KIAA1324, NOA1, RAB11FIP1, CDKN1A, PPP1R7, UCP2, BCL2L1, CFAP46, GRHL2, SINHCAF, LRBA, DNAH5, SPAG1, ESR1, H3F3B, SRI, FLNB, EVI5, HSF1, SRD5A2, NFE2L2, WNT9A, UGDH, C2orf88, SOD1, STK33, PAIP2, CD46, CCDC170, CEP19, SDC4, NQO1, DYDC1, EFHC2, KCNC3, RMND5A, POLR2B, CD47
================================================================================
pred_cell_type = Endothelial Cells | Top100 genes
AQP1, PTPRB, EPAS1, FLT1, SOCS3, SLCO2A1, PECAM1, TGFBR2, PLVAP, CD93, SHANK3, EGFL7, CD34, ENG, TIMP3, CLDN5, TMEM255B, EMP1, TIE1, HSPG2, CCL14, KDR, CAVIN2, COL4A1, MYH9, CDKN1A, PLPP3, CALCRL, MMRN2, PTPRM, SPTBN1, NOSTRIN, SLC2A3, PODXL, ITGA6, ELK3, FLT4, S1PR1, NES, CLEC14A, GIMAP4, CXCL2, TFPI, NRN1, NOS3, TNS2, HERC2, ROBO4, SLC9A3R2, MALL, C7, THBD, COL4A2, GIMAP8, SHC1, TEK, ESM1, DUSP6, PDGFB, CDH5, ENPP2, ENTPD1, DOCK9, PCDH17, VEGFC, ADGRL4, JAG1, JAM2, KLF4, EGR3, PDE2A, SELP, FZD4, LRRC32, TSPAN7, NOTCH1, IL3RA, SH2D3C, SULF2, IL33, DUSP5, TCF4, AFAP1L2, ABI3, MSN, SOX18, TGFBR3, SOCS2, BMPR2, FAM107A, MRTFB, SH3BGRL2, TINAGL1, TSPAN13, TCIM, MCAM, GIMAP5, INHBB, ECE1, APLNR
================================================================================
pred_cell_type = Fallopian Tube Epithelium | Top100 genes
EEF1G, AC007906.2, H3F3B, KCNC3, CLDN1, PAX8, LENG8, MUC1, PFKFB3, GAS6, BCAM, EHF, C2orf88, CCND2, ESR1, IMPDH2, TBX2, WDR77, PKP2, FAM107A, ATF3, ERP27, OGT, PLXNB1, SLC40A1, THSD4, CD22, S100A1, CHD3, SORL1, SOX9, FBL, PLXNB2, PIK3R1, PAX2, PDCD4, PODXL, EMX2, NONO, NTN4, IGBP1, MYOF, SCNN1A, GTF2I, FMOD, ADCYAP1R1, KCNMA1, SOST, FGFR2, ST14, WDR72, PHB2, MLPH, ATP5F1C, KAT2A, SF3B1, TMEM98, PCBD1, RAI2, LGR5, CTNND1, BMP7, POGZ, MTA1, MSLN, HSPB1, GREB1, PHF10, PTPRF, HOXB6, PAM, RAB11FIP1, DEPTOR, TNFRSF14, CCDC6, GUSB, APOBEC3C, MAP3K1, AR, ITGB4, CELSR2, HADHA, RERG, C4B, NPR1, PNOC, SDHA, TMEM45B, MPP1, FOLR1, NR4A1, CDH1, DPP4, HNF1B, SIAE, ABI3BP, PPP1R1B, MCTP2, CCT3, TDRD3
================================================================================
pred_cell_type = Granulosa Cells | Top100 genes
TRIM29, SLC40A1, TNS4, H3F3B, DSC3, HSPB1, LCN2, PLAT, CDKN1A, SOCS3, NAMPT, ANXA8, FGFR3, ATF3, EHF, AVPI1, AHR, KLF5, TNFRSF21, MALL, CTNND1, CLDN1, AC007906.2, PROM2, CD44, HES1, TNFSF10, NR4A1, SGK1, NTN4, RAB11FIP1, PKM, EPHA2, ITGB6, NFE2L2, AGR2, GABRP, ITGB4, ATP1A1, NECTIN1, LAMB3, CYP24A1, DUSP5, HIF1A, MYOF, F3, CCND1, BARX2, PTPRF, TIPARP, ANXA2, EGFR, GSTM1, MAFF, CD82, DDR1, TGM1, HBEGF, IRF1, SMAD3, WNT5A, IFI16, GPRC5C, PLXNB2, SEMA4B, MAOA, DDX3X, ALOX5, PLEC, TC2N, CYP3A5, TRAF4, SLC2A1, MEIS2, TP63, XBP1, GRHL2, BIRC3, MET, DSG3, C4B, MUC1, SH3BGRL2, F11R, IFNGR1, HK1, SDC4, HOXA5, DST, FAT2, JAG1, FOSL1, FOXC1, PIK3R1, SCPEP1, ITGA6, RAC1, MUC4, CA12, ERBB3
================================================================================
pred_cell_type = Inflammatory Tumor Cells | Top100 genes
MX1, STAT1, IFI44L, IFIT1, IFIT3, SAMD9, GBP1, PLSCR1, DDX58, CD47, OAS3, ADAR, DNAJA1, CXCL10, UCHL1, CP, IFIT2, LAPTM4B, IFI16, RSAD2, YWHAZ, APOL1, PKM, SP110, IFI35, LCN2, HSPD1, HSPE1, SAMHD1, PTGES3, SOD1, MUC16, HSPA8, APOL2, IFIH1, TNFSF10, EIF4G1, HNRNPD, CFB, HMGA1, OAS1, EIF4A1, AP2M1, THEMIS2, PRKCI, EIF2AK2, ESR1, CDV3, EPCAM, RPN1, PLXNB1, HNRNPM, DHCR24, IFT57, AMOTL2, APOL6, NDRG1, RAB7A, XPR1, ADAMTS1, TP53, SULF1, PDIA3, HSPA9, C1QBP, SRSF1, RBBP8, STAU1, LAMP3, CCT8, BCAM, TAP1, PSMD3, IL1RAP, APP, IFITM1, XPO1, NME1, IRF1, CNP, SPATS2L, RBCK1, CTNNB1, MLF1, TOP1, CSNK2A1, ATF4, SMARCC1, AHCY, ATP5F1A, PABPC1L, KLK6, KPNB1, LRATD2, YES1, ELAVL1, CCT3, CLIC4, DSG2, CBX3
================================================================================
pred_cell_type = MT-High, Jun+/Fos+ Tumor Cells | Top100 genes
EEF1G, LAPTM4B, CD47, HSPD1, IMPDH2, FBL, YWHAZ, PRKCI, AHCY, CP, HMGA1, HNRNPD, EPCAM, C2orf88, EIF4A1, UCHL1, SOD1, TMEM123, ESR1, PKM, HSPE1, MLF1, ATP5F1A, IFT57, EIF4G1, CCT8, PEG10, CCT3, TOP2B, APP, C1QBP, LCN2, SMARCC1, PTGES3, PCNA, RBM38, CDKN2A, NIPSNAP1, CTNNB1, TP53, PHB2, CDV3, SOX9, NME1, MAL2, PABPC4, AP2M1, SOX17, HSPA9, NOA1, RAB7A, RUVBL1, LTA4H, LRRTM1, CD200, CBX3, TMEM97, DHCR24, FZD3, NDRG1, CHODL, LRPPRC, GALNT4, ADNP, PRKDC, PIK3R2, MCM2, POLR2B, STAU1, SQLE, EMX2, RBBP8, DSG2, KIAA1324, CMSS1, CSNK2A1, RMND5A, FOXJ1, CCT2, TOP1MT, RMND1, LRATD2, KPNB1, RRS1, STMN1, APEX1, GMPS, SPECC1, TFAP2A, PARP1, PTDSS1, MECOM, PTPN11, CSE1L, NUDT21, UNG, MCCC1, CDH1, ERBB3, DKC1
================================================================================
pred_cell_type = Macrophages | Top100 genes
C1QC, MS4A6A, AIF1, FCGR2A, CD14, ITGB2, STAB1, FCGBP, MSR1, SGK1, SLCO2B1, C5AR1, NAMPT, CYBB, CD163, LIPA, MNDA, MAFB, SIRPA, CD4, CD68, LAIR1, ZEB2, LGMN, CIITA, PLAUR, CSF1R, IFNGR1, HSPA6, GPR34, F13A1, CTSL, MRC1, CLEC5A, FCGR2B, SAMHD1, MPP1, ARRB2, GRB2, SPI1, FCGR3B, HIF1A, MEF2A, CTSC, AP1B1, CCL2, PFKFB3, TLR4, CD300A, GIMAP4, DAB2, TLR2, TGFBR1, SORL1, MAN2B1, SIGLEC1, TNFRSF11A, ANXA5, TLR7, IL10RA, GRN, IL13RA1, CD44, ITGA4, ALOX5, CX3CR1, CD83, CCR1, TLR1, HTRA1, CXCR4, VSIG4, CD93, HMOX1, CLEC7A, HAVCR2, MAF, ITGAM, FCGR3A, PLTP, C3AR1, IKZF1, BTK, LILRB1, SLC7A7, DPYD, FPR1, ABCA1, CXCL16, TREM2, ITGAX, LPAR6, ADAM8, ABI3, HNMT, SLC2A3, LILRB4, MSN, TNFRSF1B, PLEKHO1
================================================================================
pred_cell_type = Malignant Cells Lining Cyst | Top100 genes
CLDN1, CCDC80, PROCR, PRG4, C4B, DSC3, CFB, CALB2, CCN1, RGS4, BNC1, KDR, PDPN, IL1R1, PIM1, ANXA2, MAF, AQP1, CPA4, SLC39A8, CCND2, TUBB, ANXA5, CCN2, PLS3, ITGA3, ANXA8, ITGB5, DCN, SLC40A1, PTGER3, SIRPA, TMEM98, HTRA1, VGLL3, PODXL, RAC1, POSTN, SEMA3C, BGN, PLAT, GATA6, HIF1A, HPCAL1, TSPAN3, SERPINH1, AOX1, GAS6, DAP, CXCL12, HSPB1, DAPK1, NAMPT, VCAM1, CFH, DUSP6, MYLK, LTBP2, HSPA8, P4HB, CKAP4, MMP14, CANX, RAB11FIP1, IFI16, H3F3B, TNFRSF21, TMEM14C, GXYLT2, PROS1, COL5A1, GTF2I, INMT, CTTN, SGK1, TIMP2, FGF1, MET, PEA15, LGALS8, GLS, SHC1, NPNT, SPATS2L, TPBG, PIEZO2, PLXNB2, SOCS3, CCDC71L, SULF1, RTN4, SULF2, ABCA8, THBS2, NECTIN2, ANXA3, NFE2L2, FMOD, LRP2, ATF3
================================================================================
pred_cell_type = Pericytes | Top100 genes
MYH11, C11orf96, MYL9, HEYL, BGN, PDGFRB, COL4A1, EPAS1, ITGA7, COL4A2, TIMP3, NOTCH3, MCAM, SEPTIN4, AVPR1A, AOC3, JAG1, TBX2, TINAGL1, ADGRA2, THY1, CDKN1A, SDC2, EBF1, ENPEP, PDGFA, ADAMTS4, ANO1, NR4A1, ITGA1, STEAP4, SYNPO2, CCDC102B, PLN, MYO1B, ZEB2, COL18A1, RGS5, ABCC9, C4B, S1PR3, TNC, EDNRA, PDE5A, POSTN, KCNJ8, MYLK, MFGE8, NES, TFPI, PTP4A3, CSPG4, LRRC32, PARM1, PGF, TRPC6, MYH9, SEPTIN5, CNN1, MAP1B, AQP1, TNS2, CACNA1H, C1QTNF1, SNAI2, SYTL2, FILIP1L, CD36, MAP3K7CL, ADAMTS1, NREP, PHLDB2, GUCY1A2, GJC1, SOD3, SLC2A3, RASD1, PRRX1, SCN4B, AFAP1L2, ADAMTS12, MAPRE2, PCDH18, RCAN2, FLT1, AXL, COL5A1, NDRG2, RBPMS, TGFB3, PLS3, NFATC4, C7, CAV2, PLXDC1, SMOC2, EGFL6, DST, CCL2, SLIT3
================================================================================
pred_cell_type = Proliferative Tumor Cells | Top100 genes
UCHL1, HNRNPD, SMC4, CD47, LAPTM4B, HMGA1, TPX2, BIRC5, HSPD1, AHCY, TOP2A, PRKCI, MYBL2, PTGES3, EPCAM, ESR1, HNRNPM, HSPE1, PLXNB1, PCNA, SRSF1, TUBB, STMN1, PKM, YWHAZ, EIF4G1, SFPQ, SOD1, ILF3, PABPC1L, RPN1, TYMS, CBX3, FBL, ADAMTS1, CSE1L, IRAK1, CP, SLC2A4RG, CIP2A, CENPF, CYP4B1, CDK1, ELAVL1, CCNE1, EIF4A1, AP2M1, ANP32E, SOX17, XPO1, GMPS, TOPBP1, PRKDC, ATP5F1A, CBX5, EEF1G, TMEM97, TP53, TOP2B, TFRC, CCT8, CSNK2A1, KPNB1, USP53, ALCAM, SMARCC1, RBBP4, SDHA, NMU, DEK, UBE2C, RBMX, SPIN1, MCM4, F11R, HSPA8, BCAM, DNAJA1, RUVBL1, KPNA2, GANAB, IFT57, C1QBP, IL1RAP, NME1, CCT3, CD200, MLF1, CDKN1B, IMPDH2, HSPA9, POLR2B, UQCC1, DNAJC9, FZD3, AURKB, MCM2, RGL3, MAL2, IGFBP2
================================================================================
pred_cell_type = Smooth Muscle Cells | Top100 genes
MYH11, CNN1, C11orf96, MYL9, MYLK, CSRP1, NR4A1, ATF3, CDKN1A, POSTN, PLN, AOC3, PLPP3, SYNPO2, SMTN, CCN1, HSPB1, ITGA7, SYNM, MAOB, C4B, GRIA2, SVIL, SLMAP, MEIS3, DST, HSPB8, JPH2, OSR2, CCND2, NCAM1, SMOC2, ABCC9, GATA2, KCNMA1, FHL1, LTBP1, PDE5A, TACC2, FLNC, GUCY1A2, GABBR1, ADAMTS1, PTGIS, ANO1, PIM1, IGSF9B, EDNRA, CBX7, RBPMS, TGFBR3, CACNA1H, BNC2, FGF7, CA12, CDH3, GPRASP1, COL4A2, TGFB1I1, AQP1, RERG, TRAF5, ADAM33, PTGFRN, ITPR1, ABI3BP, KCNMB1, HOXA5, BCHE, PLIN4, PRRT2, MAP3K20, MFGE8, MAFF, ADAMTSL3, ALDH1B1, FILIP1L, FGFR1, PER1, ACTN1, MID2, PALLD, ENTPD1, RSPO3, CMYA5, HPGD, COL4A5, CPXM2, DMPK, FGF2, CLDN1, NEXN, ILK, LDB3, CNTNAP1, ITGB3, TSPAN18, ENPP1, SOD3, PKD1
================================================================================
pred_cell_type = Stromal Associated Fibroblasts | Top100 genes
C7, DCN, ABCA10, CCN1, ABCA8, NR4A1, PLPP3, TNXB, CCDC80, KLF4, LUM, SOCS3, PAMR1, SRPX, PDGFRA, NAMPT, THBS1, F3, NBL1, DPT, IL6, ABI3BP, CTSK, LRP1, PIM1, CXCL12, TIMP3, CCDC71L, ADAMTS1, PDGFRB, AOX1, SFRP1, C4B, MAFF, CD34, SLC2A3, VLDLR, CDKN1A, ADM, EMP1, SVEP1, C11orf96, SFRP4, FGF7, ADAM33, EGR3, SNAI2, CXCL2, SLIT2, EBF1, H3F3B, TNS2, SLIT3, OSR2, NGFR, PRRX1, TFPI, OLFML3, RBMS3, ADGRA2, COLEC12, NR4A3, FGFR1, ADAMTSL3, PI16, HOXA5, TWIST2, FBLN5, PTGS2, CYP1B1, HEYL, PER1, SLC40A1, SMOC2, MFGE8, ANGPTL2, NDRG2, ADAMTS4, CD248, SCN7A, GATA2, THBS2, FAIM2, CCL2, PTGIS, PCDH18, PMP22, TMEM100, FRMD6, RASD1, CCND2, INMT, SDC2, PID1, MAN1A1, GABBR1, NCAM1, TNFRSF14, SERPINA3, TNFSF14
================================================================================
pred_cell_type = T & NK Cells | Top100 genes
TRAC, CD52, CD8A, CXCR4, TRBC1, IL7R, CD3E, CD3D, CD2, IKZF1, CD96, TBC1D10C, GZMA, IL2RG, GZMH, ITGAL, KLRK1, CTSW, CD44, CD247, SH2D1A, GIMAP4, CCND2, IKZF3, ITGB2, GPR174, SEPTIN6, SLAMF7, IL16, RASSF5, CTLA4, NLRC5, GBP5, IRF1, IL2RB, CD3G, ACAP1, TRAF3IP3, CD6, GZMK, BIRC3, ITGA4, ZAP70, BCL2, KLRG1, APOBEC3C, SELPLG, RASGRP1, PIM2, P2RX5, STAT4, KLRB1, LCK, CD7, CYLD, IL10RA, CD5, BCL11B, LAT, APOBEC3G, C1QC, CST7, ITM2A, KCNA3, GIMAP5, ADGRE5, MS4A1, PIK3CG, SLAMF6, APOL6, EVI2B, PTGER4, SEMA4D, SP140, TENT5C, WAS, ITK, CD28, ETS1, FLT3LG, SPN, TNFAIP8, PTPN22, PIM1, CARD11, MDFIC, ZNF683, TIGIT, ADAM8, PTGDR, CD38, PRDM1, SH2D2A, PRKACB, CYFIP2, IQGAP2, CXCR6, PRKCH, FYN, KLRC1
================================================================================
pred_cell_type = Tumor Associated Fibroblasts | Top100 genes
BGN, LUM, COL5A1, DCN, MMP14, CTHRC1, POSTN, COL5A2, TIMP3, CCN1, CCN2, SERPINH1, LOXL1, THBS2, COL11A1, THBS1, CDH11, TIMP2, THY1, NBL1, PLAU, CXCL12, LTBP2, INHBA, SULF1, GXYLT2, AEBP1, SERPINE1, ITGB5, SFRP4, MRC2, IGFBP2, ADAMTS14, MYH9, FAP, COL10A1, LOX, CKAP4, CCN4, ANXA5, COL4A1, MMP11, PTGIS, SEMA3C, HTRA1, GAS6, SPATS2L, MYL9, CD99, ANXA2, SNAI2, FMOD, OLFML3, ADAM12, HOPX, CCDC80, FSTL1, LRP1, PDGFRA, ECM1, COL4A2, RUNX1, PRRX1, P4HB, PIM1, PTK7, CTSK, NREP, MICAL2, CFH, SULF2, DAP, TPBG, MAP4K4, TUBB, FILIP1L, RUNX2, ADAMTS2, PDGFRB, SLC2A3, PDPN, ADAMTS12, VCAM1, SHC1, CDKN1A, PTGFRN, PALLD, DST, TSPAN9, TGFB3, TMEM45A, C1QTNF6, DSC3, ADGRA2, FZD1, C4B, PLXNB2, SDC2, CCND2, PLS3
================================================================================
pred_cell_type = Tumor Cells | Top100 genes
PLXNB1, EEF1G, CP, CD47, LAPTM4B, IMPDH2, ESR1, PRKCI, AHCY, MUC16, RGL3, PABPC1L, FBL, HNRNPD, APP, HSPD1, BCAM, TMEM123, EPCAM, YWHAZ, CYP4B1, UCHL1, CTNNB1, EIF4A1, EIF4G1, CCT3, HNRNPM, SDHA, UQCC1, CSNK2A1, SRSF1, SPIN1, INPPL1, SMARCC1, CBX3, CD200, PKM, XPO1, ATP5F1A, ILF3, SOD1, CCT8, RBM12, F11R, TOP2B, HMGA1, KCNC3, C2orf88, HOXB4, NIPSNAP1, MTAP, AMOTL2, PHB2, IRAK1, PABPC4, DNAJC10, SFPQ, PTPRF, RBM38, DSG2, MEIS1, THSD4, PIK3R2, LRATD2, PTGES3, NDRG1, MAL2, ADNP, MLF1, CHD3, DDR1, BCL2L1, IL1R1, HSPE1, PLXNB2, PAWR, RBMX, SLC2A4RG, RPN1, TP53, PRKDC, APEX1, SULF1, ELAVL1, DHCR24, CPSF6, POLR2B, PAX8, SINHCAF, SOST, SF3B1, CDV3, AP2M1, ANKRD17, SOX9, EMX2, MAP4, NEPRO, KPNB1, C1QBP
================================================================================
pred_cell_type = VEGFA+ Tumor Cells | Top100 genes
CD47, YWHAZ, NDRG1, LAPTM4B, TFPI2, AMOTL2, EEF1G, LCN2, HSPD1, PRKCI, PKM, VEGFA, APP, CDV3, SLC2A1, PTGES3, CP, EIF4G1, HSPE1, EIF4A1, MLF1, EPCAM, FBL, AHCY, TFRC, IMPDH2, ALCAM, CCT8, SPECC1, HMGA1, IL1RAP, IFT57, TMEM123, DSG2, CSNK2A1, HNRNPD, ADAMTS1, DNAJA1, DHCR24, RAB7A, HSPA9, STAU1, ATP5F1A, P4HA1, YES1, ATP1A1, BCL2L1, SMARCC1, MUC16, CCT3, ANKRD33B, AP2M1, VDAC2, CTNNB1, BRD4, SOD1, CXADR, TP53, HNRNPM, POLR2B, KLK6, SINHCAF, RAC1, RPN1, UCHL1, LRATD2, TCP1, GMPS, DPM1, KPNB1, CCT2, MAL2, SDC4, SMAD2, ARL8B, HSPA8, BCAM, ELAVL1, ARF1, CBX3, GADD45A, ATF4, APOL1, ITCH, C1QBP, SPIN1, PTPN2, MMADHC, CDH6, NEPRO, CCT6A, PAK2, LRPPRC, NAMPT, PTPN11, KHDRBS1, KIF5B, ESR1, RBM38, SYNCRIP
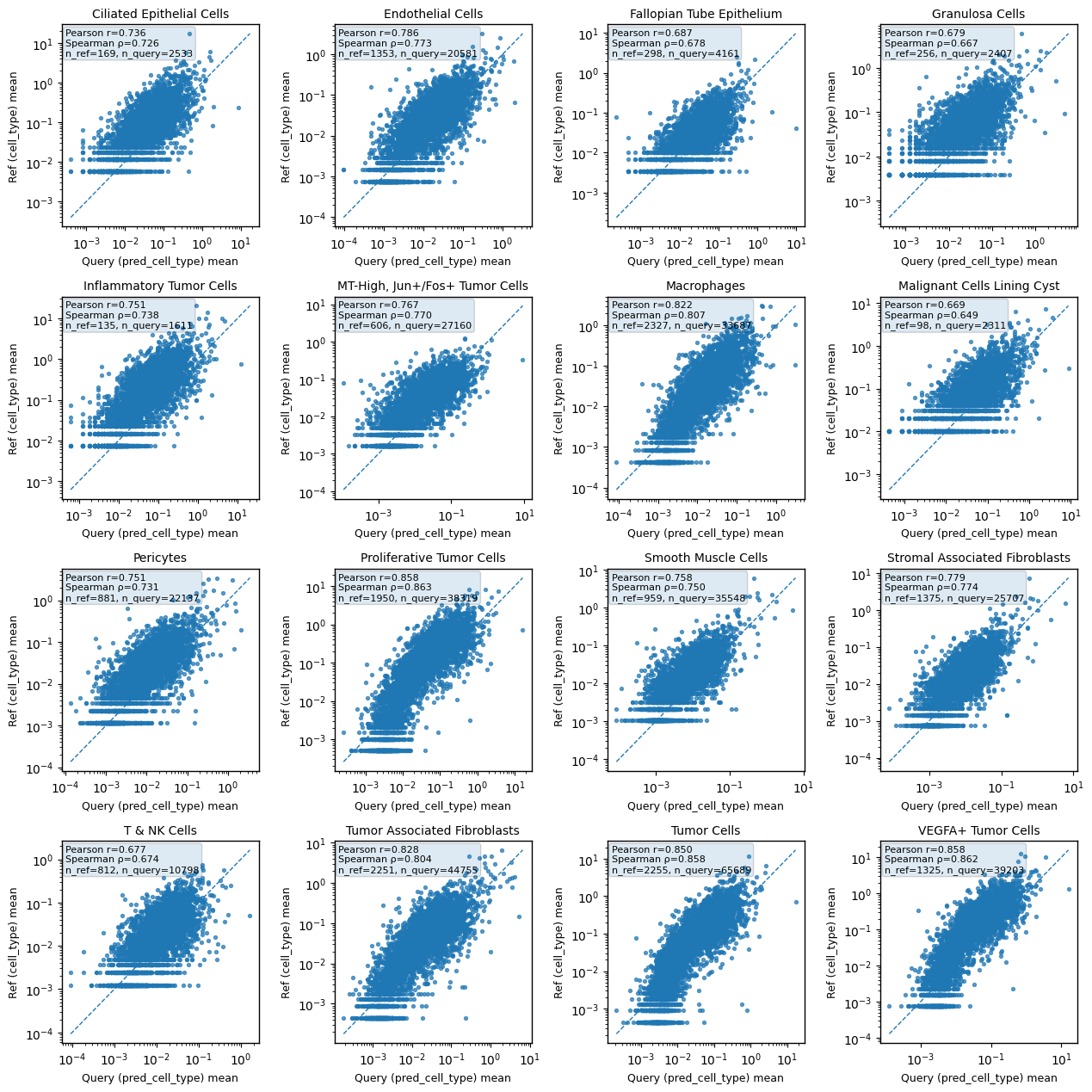
Visualize (correlation)#
Compare reference vs predicted cell-type gene mean profiles.
import scbiot as scb
fig, corr_df, means_ref_df, means_qry_df = scb.pl.celltype_gene_mean_correlation(
adata_ref,
adata_query,
ref_group_key="cell_type",
query_group_key="pred_cell_type",
layer="counts",
use_raw=False,
min_cells=30,
ncols=4,
)
# print(corr_df.head(10))
# fig.savefig("celltype_gene_mean_correlations.png", dpi=200, bbox_inches="tight")
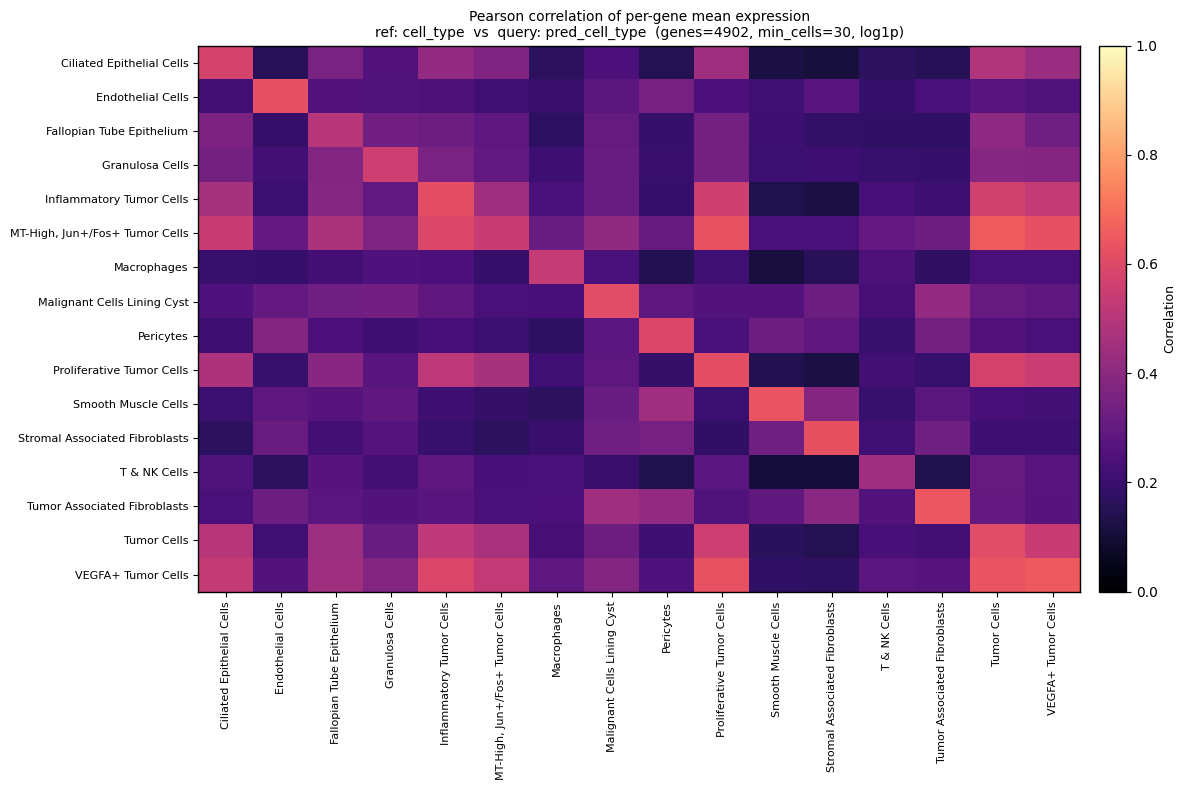
# same as above, different visualization as a heatmap
fig, ax, corr_df, means_ref_df, means_qry_df = scb.pl.celltype_predtype_mean_corr_heatmap(
adata_ref,
adata_query,
ref_group_key="cell_type",
query_group_key="pred_cell_type",
layer="counts",
use_raw=False,
min_cells=30,
transform="log1p",
method="pearson",
figsize=(12, 8),
vmin=0.0,
vmax=1.0,
)
adata_query.obs['pred_cell_type'].value_counts()
pred_cell_type
Tumor Cells 65689
Tumor Associated Fibroblasts 44755
VEGFA+ Tumor Cells 39203
Proliferative Tumor Cells 38319
Smooth Muscle Cells 35548
Macrophages 33687
MT-High, Jun+/Fos+ Tumor Cells 27160
Stromal Associated Fibroblasts 25707
Pericytes 22137
Endothelial Cells 20581
T & NK Cells 10798
Fallopian Tube Epithelium 4161
Ciliated Epithelial Cells 2533
Granulosa Cells 2407
Malignant Cells Lining Cyst 2311
Inflammatory Tumor Cells 1611
Name: count, dtype: int64