5. Unpaired Multiomics Integration#
Source: https://scib-metrics.readthedocs.io/en/latest/notebooks/lung_example.html
import warnings
import os
from pathlib import Path
import matplotlib.pyplot as plt
import matplotlib as mpl
import scanpy as sc
# import rapids_singlecell as rsc
import anndata
import numpy as np
import pandas as pd
from sklearn.metrics import normalized_mutual_info_score
import scbiot as scb
from scbiot.utils import set_seed
warnings.filterwarnings("ignore")
set_seed(42)
dir = Path(os.environ.get("SCBIOT_EXAMPLES_PATH", Path.cwd()))
print(dir)
print(dir.parent)
verbose = True
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/_utils/__init__.py:33: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
from anndata import __version__ as anndata_version
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/__init__.py:24: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
if Version(anndata.__version__) >= Version("0.11.0rc2"):
/home/figo/software/python_libs/scbiot/.venv/lib/python3.12/site-packages/scanpy/readwrite.py:16: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('anndata')` instead.
if Version(anndata.__version__) >= Version("0.11.0rc2"):
scbiot version 1.1.8
Random seed set as 42
/home/figo/software/python_libs/scbiot/examples
/home/figo/software/python_libs/scbiot
Load#
Read the RNA and ATAC inputs from disk.
adata_gex_path = f'{dir}/inputs/Yao-2021-RNA.h5ad'
adata_atac_path = f'{dir}/inputs/Yao-2021-ATAC.h5ad'
adata_gex = sc.read(
adata_gex_path,
backup_url="https://figshare.com/ndownloader/files/59742656",
)
adata_atac = sc.read(
adata_atac_path,
backup_url="https://figshare.com/ndownloader/files/59742647",
)
Build reference UMAP if want to map embeddings to reference#
Preprocess#
Filter peaks, build gene activity, and align metadata across modalities.
# 0) ATAC preprocessing (peak filtering -> LSI -> GA + smoothing)
# figshare link: https://figshare.com/ndownloader/files/59742641
gtf_file = f"{dir}/inputs/gencode.vM25.chr_patch_hapl_scaff.annotation.gtf.gz"
adata_ga = scb.pp.create_gene_activity(adata_atac, adata_gex, gtf_file=gtf_file, verbose=True)
Removed 19,172 promoter-proximal peaks (2000bp upstream / 500bp downstream). Remaining: 129,642
Running Iterative LSI iteration 1 ...
Running Iterative LSI iteration 2 ...
[GA] Kept 22,358/56,262 genes by biotype ['protein_coding', 'lncRNA']
[GA] Peaks contigs: 21; Genes contigs: 108; Common: 21
[GA] Using gene field: gene_name
[GA] Built GA with shape (54844, 15547) (cells × genes) from 148,814 peaks.
[names] Harmonized symbols; overlaps (case-insensitive): 15,185
import anndata
# adata_gex.write_h5ad('adata_gex.h5ad')
# adata_ga.write_h5ad('adata_ga.h5ad')
# adata_atac.write_h5ad('adata_atac.h5ad')
adata_gex = anndata.read_h5ad('adata_gex.h5ad')
adata_ga = anndata.read_h5ad('adata_ga.h5ad')
adata_atac = anndata.read_h5ad('adata_atac.h5ad')
import numpy as np
import scipy.sparse as sp
import scanpy as sc
import anndata
import scbiot as scb
# Strore raw counts
adata_gex.layers["counts"] = adata_gex.X.copy()
adata_ref_umap = adata_gex.copy()
sc.pp.highly_variable_genes(adata_ref_umap, n_top_genes=2000, flavor="seurat_v3")
sc.pp.normalize_total(adata_ref_umap)
sc.pp.log1p(adata_ref_umap)
sc.pp.scale(adata_ref_umap)
sc.tl.pca(adata_ref_umap, n_comps=30, use_highly_variable=True)
sc.pp.neighbors(adata_ref_umap, use_rep="X_pca")
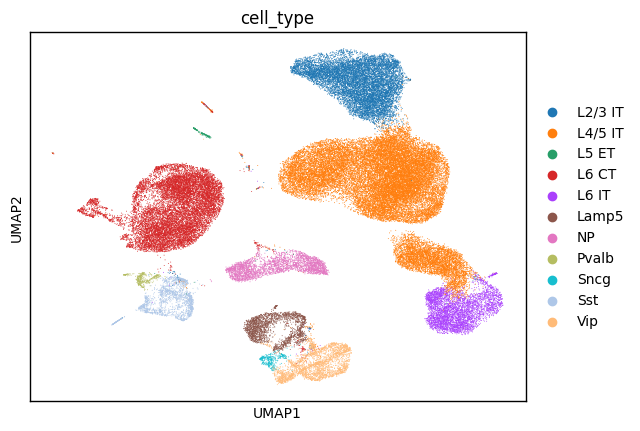
sc.tl.umap(adata_ref_umap)
sc.pl.umap(adata_ref_umap, color='cell_type')
print(adata_ref_umap)
print(adata_ref_umap.uns['umap'])
print(adata_ref_umap.uns['neighbors'])
adata_gex.obsm['X_umap'] = adata_ref_umap.obsm['X_umap']
AnnData object with n_obs × n_vars = 69727 × 27123
obs: 'x', 'cluster_label', 'subclass_label', 'class_label', 'cluster_color', 'size', 'aggr_num', 'umi.counts', 'gene.counts', 'library_id', 'tube_barcode', 'Seq_batch', 'Region', 'Lib_type', 'Gender', 'Donor', 'Amp_Name', 'Amp_Date', 'Amp_PCR_cyles', 'Lib_Name', 'Lib_Date', 'Replicate_Lib', 'Lib_PCR_cycles', 'Lib_PassFail', 'Cell_Capture', 'Lib_Cells', 'Mean_Reads_perCell', 'Median_Genes_perCell', 'Median_UMI_perCell', 'Saturation', 'Live_percent', 'Total_Cells', 'Live_Cells', 'method', 'exp_component_name', 'mapped_reads', 'unmapped_reads', 'nonconf_mapped_reads', 'total.reads', 'doublet.score', 'domain', 'protocol', 'dataset', 'cell_type'
var: 'gene_ids', 'feature_types', 'genome', 'chrom', 'chromStart', 'chromEnd', 'name', 'score', 'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount', 'blockSizes', 'blockStarts', 'gene_type', 'gene_name', 'mgi_id', 'havana_gene', 'tag', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'gene_name_orig', 'mean', 'std'
uns: 'hvg', 'log1p', 'pca', 'neighbors', 'umap', 'cell_type_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities'
{'params': {'a': 0.5830300199950147, 'b': 1.334166993228519}}
{'connectivities_key': 'connectivities', 'distances_key': 'distances', 'params': {'n_neighbors': 15, 'method': 'umap', 'random_state': 0, 'metric': 'euclidean', 'use_rep': 'X_pca'}}
Integrate#
Co-embed RNA/ATAC, run OT integration, and infer labels with supBIOT.
adata = scb.pp.coembed_pca(
adata_gex, adata_ga,
label="modality",
keys=("reference", "query"),
reference_layer="counts",
query_layer='ga_smooth',
out_key="X_shared_pca",
label_key="cell_type",
unlabeled_category="Unknown",
n_top_genes=4000,
n_components=50,
flag_outlier=True
)
[ok] L2/3 IT: n=10915 k=30 thr=14.62 out=391 (3.58%)
[ok] L4/5 IT: n=29721 k=30 thr=13.11 out=1021 (3.44%)
[ok] L5 ET: n=161 k=30 thr=22.25 out=17 (10.56%)
[ok] L6 CT: n=13361 k=30 thr=12.77 out=586 (4.39%)
[ok] L6 IT: n=4514 k=30 thr=14.72 out=194 (4.30%)
[ok] Lamp5: n=2357 k=30 thr=16.46 out=77 (3.27%)
[ok] NP: n=3147 k=30 thr=13.61 out=88 (2.80%)
[ok] Pvalb: n=368 k=30 thr=18.59 out=19 (5.16%)
[ok] Sncg: n=348 k=30 thr=21.88 out=18 (5.17%)
[ok] Sst: n=1869 k=30 thr=18.62 out=71 (3.80%)
[ok] Vip: n=2966 k=30 thr=19.4 out=118 (3.98%)
Integrate disjoint datasets with label transfer (scRNA-seq reference → snATAC-seq gene activity)#
For disjoint RNA vs GA datasets, use supervised OT and run prealignment first to reduce global mismatch.
CORAL prealignment
prealign="coral"
prealign_strength=0.5
or
OT prealignment
prealign="ot"
prealign_strength=0.8
Prealignment runs before supervised OT and helps when overlap between datasets is limited.
adata, metrics = scb.ot.integrate(
adata,
obsm_key="X_shared_pca",
batch_key="modality",
prealign='ot',
prealign_strength=0.8,
align_reference=True,
label_key="cell_type",
unlabeled_category="Unknown",
out_key="X_supbiot"
)
======== Stage1: supervised OT for label propagation ========
[prealign] OT-Gaussian enabled target=auto strength=0.8
[baseline] KNN backend=FAISS-GPU mix=0.0278 strain=0.00000
[iter 01] mix=0.033 overlap0=0.948 strain=0.00029 floor~0.600 J=0.172 best_it=1
[iter 02] mix=0.040 overlap0=0.905 strain=0.00089 floor~0.607 J=0.173 best_it=2
[iter 03] mix=0.049 overlap0=0.866 strain=0.00164 floor~0.614 J=0.176 best_it=3
[iter 04] mix=0.063 overlap0=0.819 strain=0.00269 floor~0.621 J=0.175 best_it=3
[iter 05] mix=0.064 overlap0=0.821 strain=0.00266 floor~0.629 J=0.178 best_it=5
[iter 06] mix=0.085 overlap0=0.768 strain=0.00404 floor~0.636 J=0.182 best_it=6
[iter 07] mix=0.119 overlap0=0.707 strain=0.00599 floor~0.643 J=0.199 best_it=7
[iter 08] mix=0.154 overlap0=0.644 strain=0.00840 floor~0.650 J=0.219 best_it=8
[iter 09] mix=0.178 overlap0=0.586 strain=0.01074 floor~0.657 J=0.223 best_it=9
[iter 10] mix=0.196 overlap0=0.556 strain=0.01334 floor~0.664 J=0.235 best_it=10
[iter 11] mix=0.205 overlap0=0.533 strain=0.01862 floor~0.671 J=0.226 best_it=10
[iter 12] mix=0.205 overlap0=0.534 strain=0.01881 floor~0.679 J=0.224 best_it=10
[iter 13] mix=0.205 overlap0=0.541 strain=0.02103 floor~0.686 J=0.227 best_it=10
[early stop] plateau reached.
[final] it*=10 mix=0.196 overlap0=0.556 strain=0.01334 tw=0.979
======== Stage2: Global OT for mapping query to reference ========
[align_reference] mix=0.251 overlap0=0.273 strain=0.15336 tw=0.964
Prediction of cell types with supBIOT#
When a label key is provided, supBIOT outputs two annotation fields:
pred_cell_type: the predicted cell type for each cell
pred_cell_confidence: the confidence score of the prediction
Cells with pred_cell_confidence < min_conf are considered low-confidence and will be assigned NA in pred_cell_type.
import numpy as np
# Initialize UMAP with NaNs
adata.obsm['X_umap'] = np.full((adata.n_obs, 2), np.nan)
# Mask for reference cells
mask = adata.obs['modality'] == 'reference'
# Assign UMAP coordinates
adata.obsm['X_umap'][mask.values, :] = adata_ref_umap.obsm['X_umap']
adata_gex.obs['cell_type'].value_counts()
cell_type
L4/5 IT 29721
L6 CT 13361
L2/3 IT 10915
L6 IT 4514
NP 3147
Vip 2966
Lamp5 2357
Sst 1869
Pvalb 368
Sncg 348
L5 ET 161
Name: count, dtype: int64
adata = scb.ot.supbiot(
adata,
rep_key="X_supbiot",
label_key="cell_type",
unlabeled_category="Unknown",
pred_label_key="pred_cell_type",
pred_conf_key="pred_confidence",
use_embedding_ref=True,
embedding_key="X_umap",
embedding_k=10,
embedding_weight_power=2.,
min_conf=0.0,
inplace=True,
)
Project joint embedding back to reference embedding (optional)#
Custom UMAP with Model Return
scanpy.tl.umap does not return the trained UMAP model. Use scbiot.tl.umap instead to obtain the model. Both functions produce identical UMAP coordinates.
Evaluate#
Inspect predicted labels and confidence distributions for the query cells.
adata_ga = adata[adata.obs['modality'] == 'query'].copy()
adata_gex = adata[adata.obs['modality'] == 'reference'].copy()
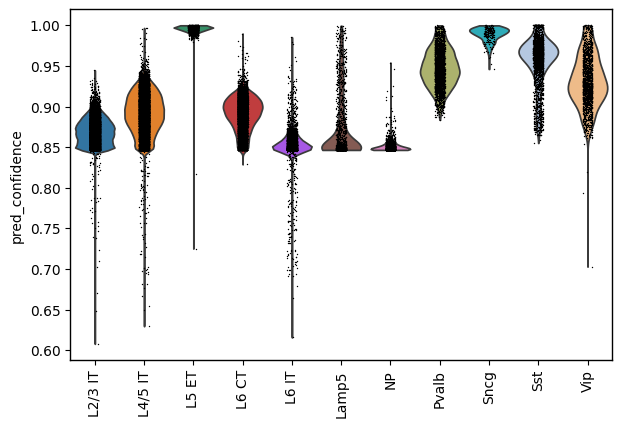
adata_ga.obs['pred_cell_type'].value_counts()
pred_cell_type
L4/5 IT 16182
L2/3 IT 15205
L6 CT 8121
L6 IT 3785
Pvalb 2751
Sst 2409
L5 ET 1819
Vip 1599
Lamp5 1428
NP 1340
Sncg 205
Name: count, dtype: int64
sc.pl.violin(adata_ga, keys="pred_confidence", groupby="pred_cell_type", rotation=90)
Visualize#
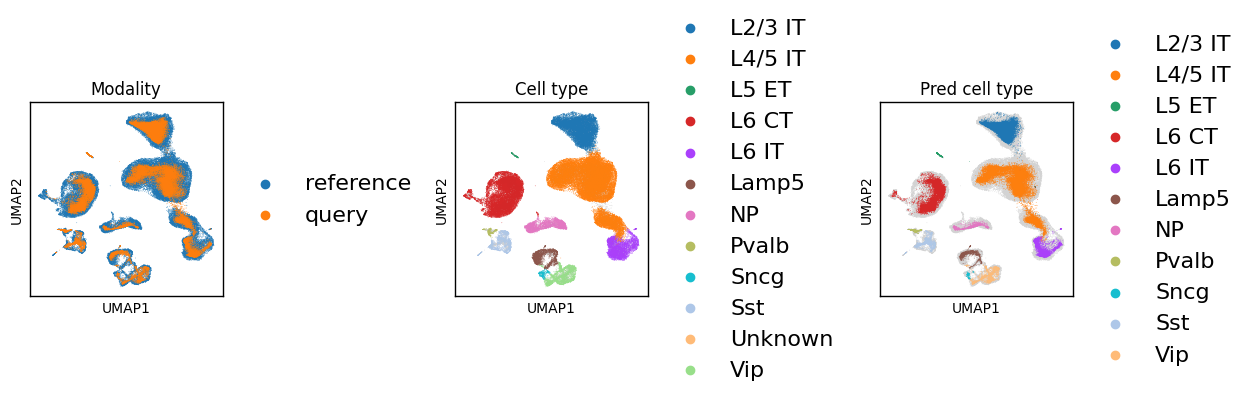
Build the UMAP on the supBIOT embedding for modality mixing checks.
# sc.pp.neighbors(adata, use_rep="X_supbiot", n_neighbors=50, metric="cosine")
# sc.tl.umap(adata, min_dist=0.3, spread=1.0, random_state=0)
import numpy as np
import pandas as pd
import matplotlib as mpl
import matplotlib.pyplot as plt
import scanpy as sc
sc.settings._vector_friendly = True
mpl.rcParams["axes.edgecolor"] = "black"
mpl.rcParams["axes.linewidth"] = 1.0
def force_border(ax):
ax.set_axis_on()
ax.set_frame_on(True)
ax.patch.set_visible(True)
for side in ax.spines.values():
side.set_visible(True)
side.set_color("black")
side.set_linewidth(1.0)
def mask_drop_unknown_na(adata, key, drop=("Unknown", "Unkown", "NA", "NaN", "None", "")):
s = adata.obs[key].astype("string")
low = s.str.strip().str.lower()
drop_low = {d.lower() for d in drop}
m = s.notna() & (~low.isin(drop_low))
return m.to_numpy(dtype=bool, na_value=False)
methods = ["X_supbiot"]
m_cell = mask_drop_unknown_na(adata, "cell_type")
m_pred = mask_drop_unknown_na(adata, "pred_cell_type")
ncols = 3 * len(methods)
fig, axes = plt.subplots(1, ncols, figsize=(4.2 * ncols, 4.2), squeeze=False)
axes = axes[0]
for i, method in enumerate(methods):
axL, axM, axR = axes[3*i], axes[3*i + 1], axes[3*i + 2]
sc.pl.embedding(
adata, basis="umap", color="modality",
frameon=True, ax=axL, show=False,
legend_loc="right margin", legend_fontsize=16, title="Modality"
)
axL.set_box_aspect(1)
axL.set_xlabel("UMAP1"); axL.set_ylabel("UMAP2")
force_border(axL)
xlim, ylim = axL.get_xlim(), axL.get_ylim()
# hide Unknown/NA cells via mask_obs (no slicing -> no view copy issues)
sc.pl.embedding(
adata, basis="umap", color="cell_type",
mask_obs=m_cell, na_in_legend=False,
frameon=True, ax=axM, show=False,
legend_loc="right margin", legend_fontsize=16, title="Cell type"
)
axM.set_xlim(xlim); axM.set_ylim(ylim)
axM.set_box_aspect(1)
axM.set_xlabel("UMAP1"); axM.set_ylabel("UMAP2")
force_border(axM)
sc.pl.embedding(
adata, basis="umap", color="pred_cell_type",
mask_obs=m_pred, na_in_legend=False,
frameon=True, ax=axR, show=False,
legend_loc="right margin", legend_fontsize=16, title="Pred cell type"
)
axR.set_xlim(xlim); axR.set_ylim(ylim)
axR.set_box_aspect(1)
axR.set_xlabel("UMAP1"); axR.set_ylabel("UMAP2")
force_border(axR)
plt.tight_layout()
Evaluate#
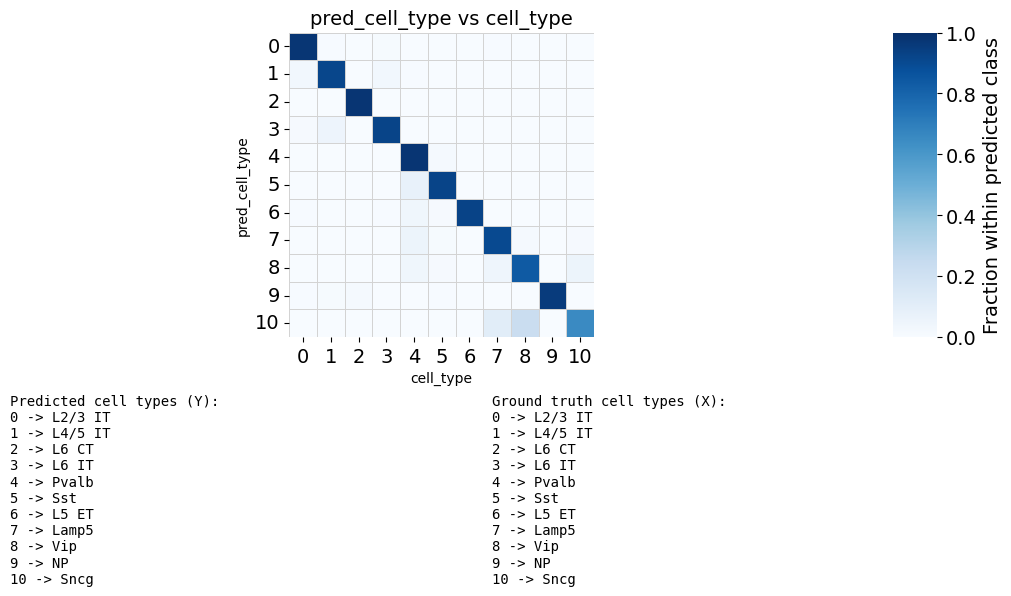
Map true labels back to ATAC cells and compute confusion/NMI metrics.
# Map true labels back
adata_ga.obs['cell_type'] = adata_atac.obs['cell_type']
fig, ax, counts_df, norm_df = scb.pl.plot_anndata_confusion(
adata_ga,
true_key="cell_type",
pred_key="pred_cell_type",
drop_unknown=False, # key line
normalize="pred",
annotate_mapping=True,
return_data=True,
)
from sklearn.metrics import normalized_mutual_info_score
y_true = adata_ga.obs["cell_type"].astype(str)
y_pred = adata_ga.obs["pred_cell_type"].astype(str)
# keep only valid rows
mask = y_true.notna() & y_pred.notna()
# (optional) ignore "unknown" labels
# mask &= (y_true != "unknown") & (y_pred != "unknown")
nmi = normalized_mutual_info_score(
y_true[mask].to_numpy(),
y_pred[mask].to_numpy(),
average_method="arithmetic",
)
print(f"NMI = {nmi:.6f}")
NMI = 0.861029